



El síndrome de Stewart-Bluefarb (SBS) o pseudosarcoma de Kaposi (PKS) es un subtipo de angiodermatitis (AD) asociado a malformaciones congénitas arteriovenosas (AVM) muy infrecuente. Presentamos un caso clínico y revisión bibliográfica de esta patología.
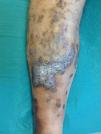
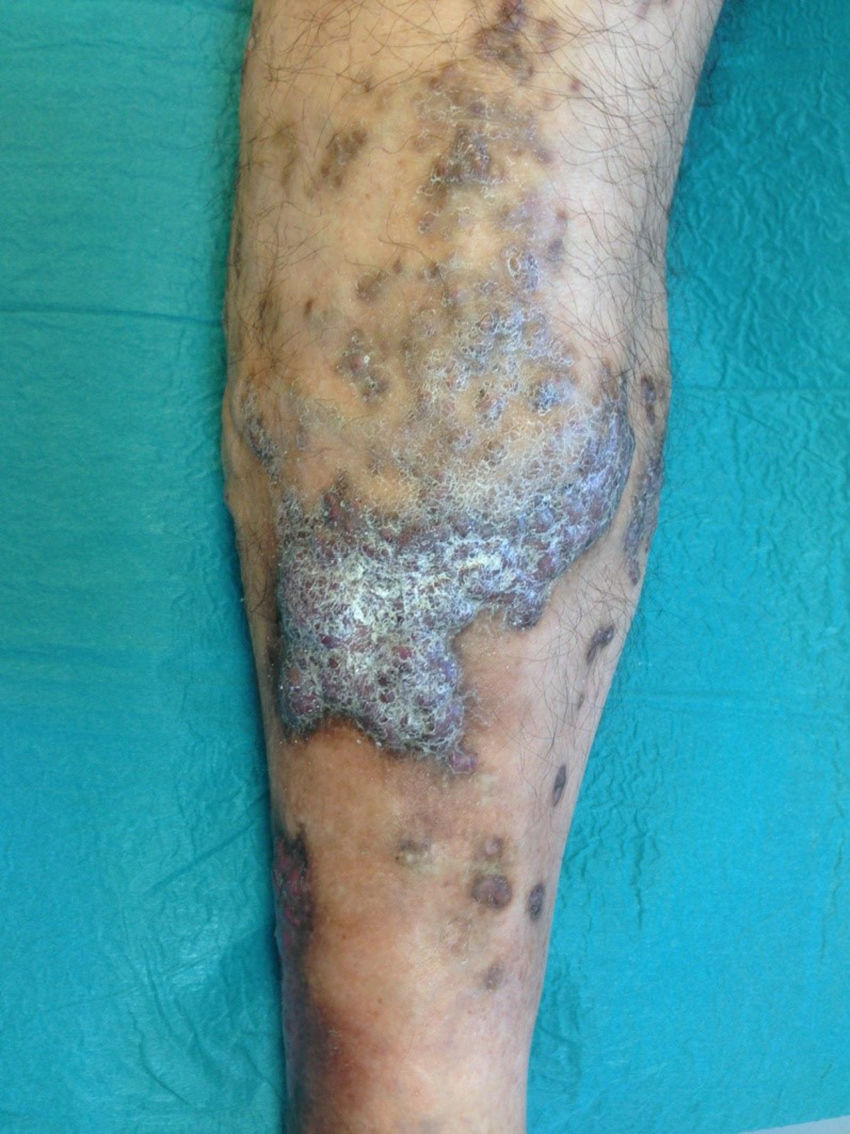
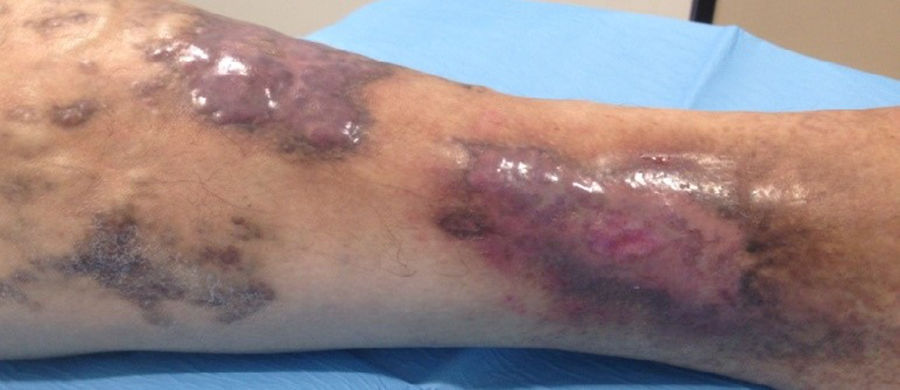
Varón de 46 años, dislipidémico y fumador activo, remitido por úlcera tórpida dolorosa desde hacía 3 meses en miembro inferior derecho. Presentaba lesiones papulosas marronáceas sobreelevadas autolimitadas y recurrentes sin filiar desde la infancia. A la exploración física, pulsos presentes simétricos y bilaterales, sin soplo ni thrill; además, lesiones tumorales sobreelevadas marrón-violáceas en región pretibial y borde externo supramaleolar con eccema perilesional y úlcera de 3×2cm con bordes irregulares, fibrina en fondo y signos leves de infección (fig. 1); sin dismetría en las extremidades inferiores. El estudio hematimétrico y bioquímico fue normal, con estudio serológico negativo. Ante la presencia de lesiones atípicas cutáneas se realizó un estudio ecografía-doppler completo de ambos miembros y una biopsia cutánea.
La histología mostró la presencia de proliferación de capilares en la dermis profunda y papilar junto con fibrosis y extravasación de hematíes, depósitos de hemosiderina y vénulas tortuosas, compatibles con SBS. La ecografía-doppler confirmó la presencia de fístulas arteriovenosas subyacentes dependientes de las arterias tibiales anterior y posterior con flujo venoso arterializado distal a la comunicación (fig. 2).
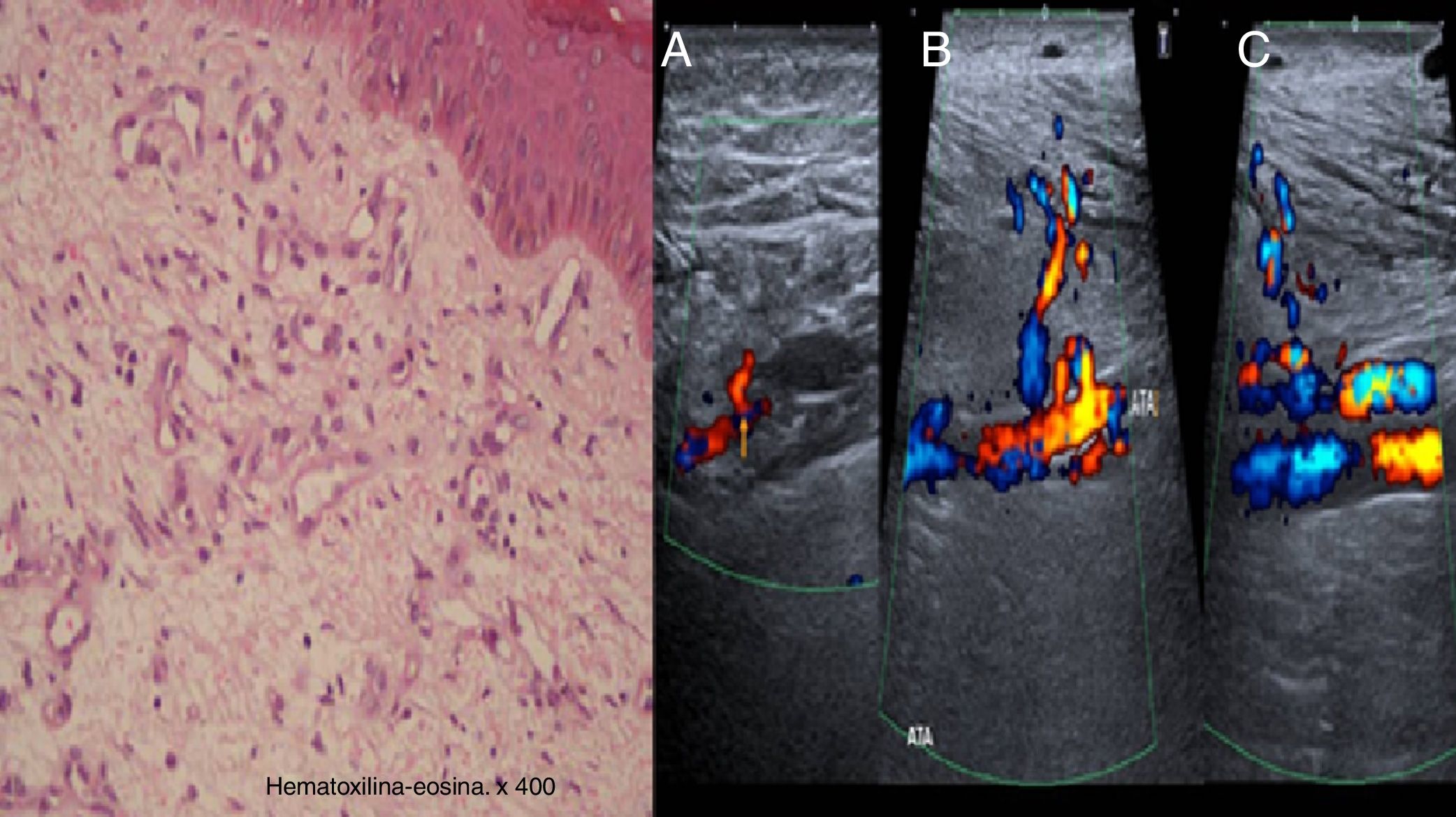
A la izquierda, imagen histológica con proliferación de capilares en dermis profunda y papilar junto con fibrosis y extravasación de hematíes, depósitos de hemosiderina y vénulas tortuosas. A la derecha, estudio eco-doppler que muestra fístula arteriovenosa dependiente de arteria tibial posterior (A), tibial anterior (B) y flujo venoso arterializado distal a nivel de la vena tibial anterior (C).
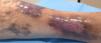
Iniciamos tratamiento conservador con antibioterapia dirigida durante 10 días tras hallar Staphylococcus aureus en el cultivo microbiológico, terapia compresiva con medias tipo calcetín sistema 2 en 1 con cremallera lateral y curas locales con un apósito lípido-coloide. A los 3 meses presentaba cicatrización de la úlcera y desaparición del dolor. Al año continúa asintomático con terapia compresiva (fig. 3).
Las AD son enfermedades angioproliferativas manifestadas mediante lesiones cutáneas clínicamente similares al sarcoma de Kaposi (KS)1-3. Consisten en una hiperplasia benigna de las estructuras vasculares preexistentes y pueden estar asociadas a AVM, como en el caso del SBS.
El SBS es una rara entidad benigna angioproliferativa, histológicamente similar al KS, publicada por primera vez por Earhart et al. en 19744, con menos de 20 casos publicados en la literatura. Afecta generalmente a pacientes varones jóvenes a partir de la segunda década de la vida5 y se caracteriza por lesiones cutáneas tipo maculopápulas de color marrón o violáceo de crecimiento progresivo y malformaciones arteriovenosas subyacentes. Se manifiesta en extremidades inferiores de forma unilateral a nivel del dorso del pie, tobillo y pantorrilla, con edema, aumento local de la temperatura, hipertrofia de tejidos blandos y varices, como consecuencia del estasis venoso2,6. Pueden complicarse con la aparición de úlceras y lesiones verrugosas1,3.
Es necesario realizar un diagnóstico diferencial con la AD de Mali, típica de pacientes ancianos con insuficiencia venosa crónica y afectación bilateral, el SK, el liquen plano, la vasculitis, y enfermedades linfoproliferativas con fístulas arteriovenosas congénitas como el síndrome de Klippel-Trenaunay-Weber o iatrogénicas en pacientes dializados o con hepatitis C4,7,8. Existen varias teorías sobre su etiopatogenia; el aumento de la presión venosa secundario a la AVM o la isquemia por robo arteriovenoso pueden suponer un estímulo para la proliferación de células endoteliales7.
La sospecha es puramente clínica (lesiones venosas atípicas, hipertrofia de partes blandas, palpación de thrill o auscultación de un soplo) con confirmación histológica, por lo que debemos familiarizarnos con esta infrecuente patología8. Histológicamente se caracteriza por proliferación de células endoteliales, angiogénesis con patrón lobular en dermis profunda y papilar y extravasación de hematíes, sin células neoplásicas fusiformes ni proliferación independiente a estructuras normales (típico del KS)1,7. La ecografía-doppler permite detectar la presencia de comunicaciones arteriovenosas2,3, aunque la arteriografía continúa siendo el gold standard1,7 para el diagnóstico y tratamiento3,5.
Actualmente el tratamiento es controvertido. La mayoría de expertos recomiendan tratamiento conservador8 y preventivo con medias de compresión y elevación del miembro si asocia edema6. El tratamiento sintomático de las complicaciones locales es crucial1, mientras que el tratamiento específico pocas veces es posible por la presencia de varias comunicaciones a nivel distal7,8. La cirugía está indicada en pacientes con impotencia funcional, dolor refractario al tratamiento, infecciones de repetición, hemorragia o descompensación cardiaca5,8. En ocasiones la amputación del miembro es la única opción9. La embolización selectiva con diferentes partículas, el láser intravenoso, la ablación con radiofrecuencia y la esclerosis guiada por ecografía pueden ser una alternativa para los casos complejos y sintomáticos5. En un caso publicado recientemente han utilizado glucosaminoglucano Cacipliq20® (heparin-sulfato mimético) en un paciente con SBS, con resolución completa de la úlcera10.
En resumen, este caso ilustra un raro síndrome de AD manifestado con una úlcera crónica dolorosa y lesiones cutáneas similares al KS que resultaron de una comunicación arteriovenosa de base. Acercamos una nueva visión para el diagnóstico mediante ecografía-doppler y se revisan las escasas y controvertidas medidas terapéuticas de esta patología en la literatura actual.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


