


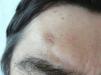

Varón de 54 años con antecedentes de HTA y dislipemia en tratamiento habitual con enalapril y simvastatina que consultó por la aparición de una lesión asintomática en la región frontal derecha de 4 meses de evolución, que había aumentado de tamaño progresivamente. No refería traumatismo previo en la zona ni haberse aplicado ningún tratamiento tópico.
Exploración físicaSe observa una placa de color piel normal, irregular, de 1 x 0,8cm de diámetro y centro ligeramente deprimido, constituida por pápulas milimétricas con distribución serpiginosa dispuestas en la periferia. No se apreciaron otras lesiones de interés en otras áreas corporales (fig. 1).
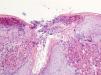
Histopatología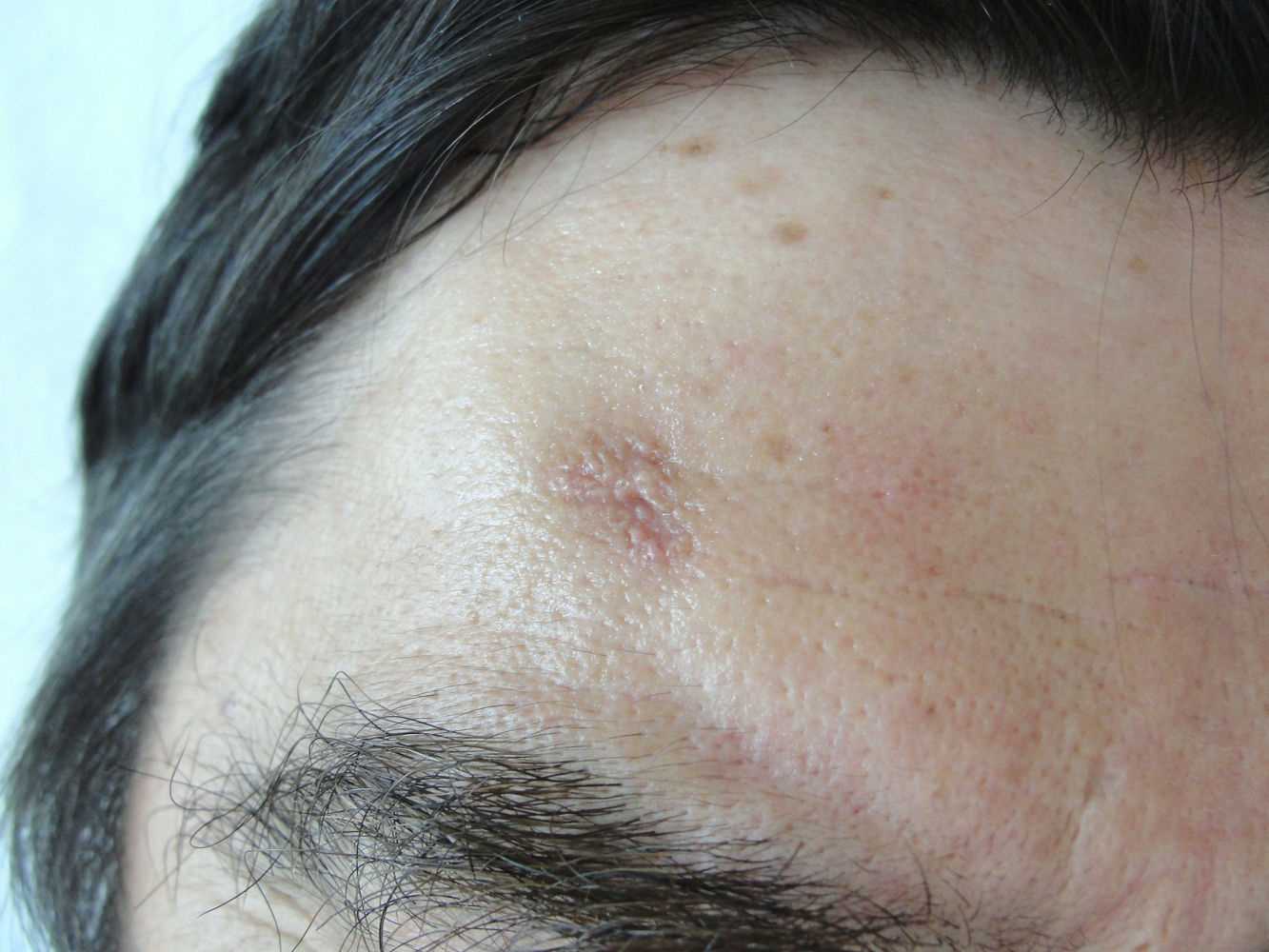
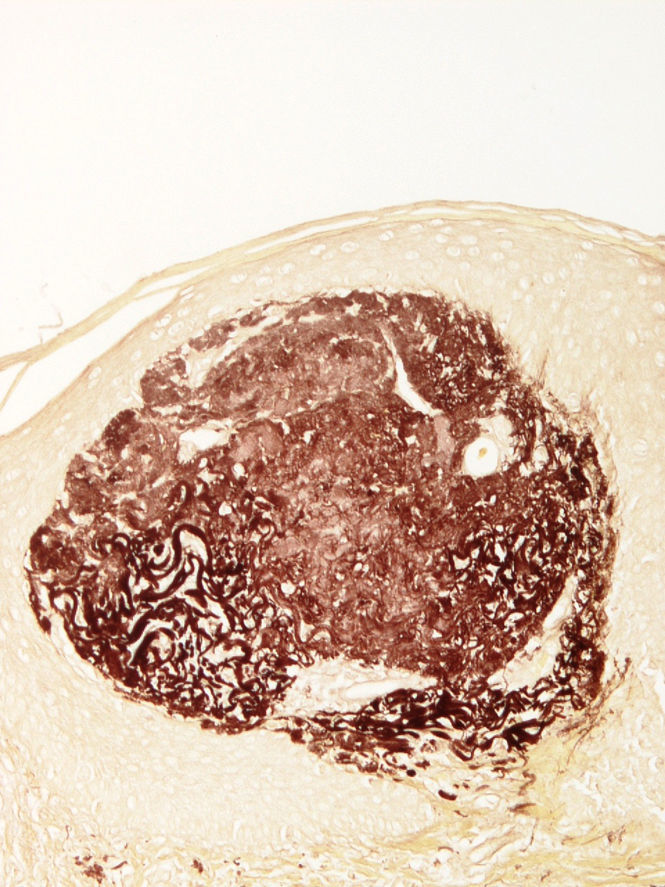
Se tomó una biopsia del borde de la lesión que mostraba en la dermis papilar superficial un cúmulo de material eosinofílico y acelular asociado a fibras elásticas gruesas e hipereosinofílicas. En la dermis adyacente se observó moderado infiltrado inflamatorio constituido por linfocitos y células plasmáticas, con frecuentes células gigantes multinucleadas. Focalmente en la epidermis supraadyacente destacaba un área de perforación de la misma, con eliminación transepidérmica del material anteriormente descrito, asociado a pequeños grupos de neutrófilos (fig. 2).
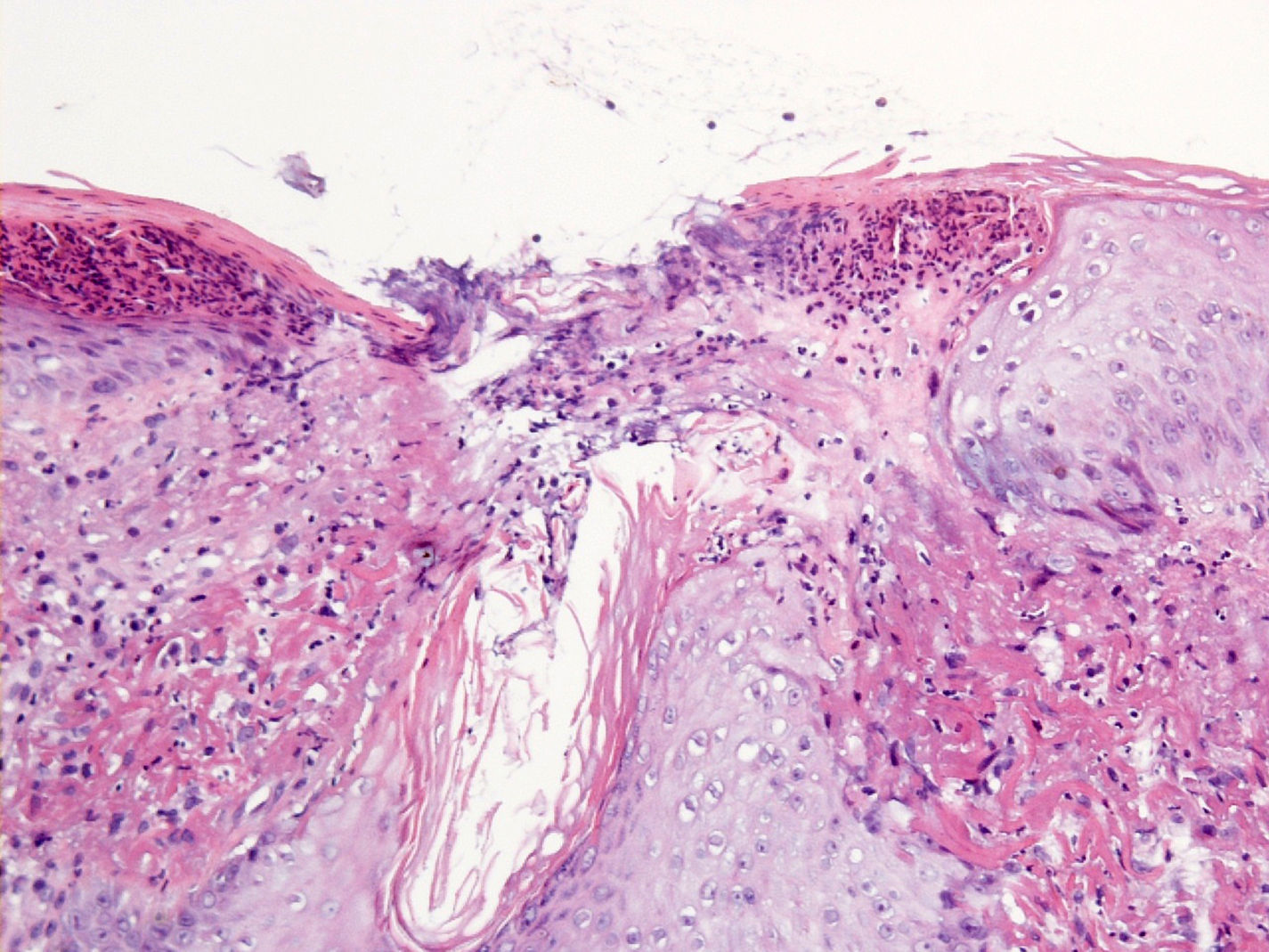
La tinción Van Gieson fue positiva solamente en las áreas con fibras elásticas alteradas, evidenciándose dicha positividad en algunas que se encontraban en la capa córnea (fig. 3).
¿Cuál es su diagnóstico?
DiagnósticoElastosis perforante serpiginosa.
Evolución y tratamientoSe realizaron dos ciclos de crioterapia en el plazo de mes y medio. A los tres meses de la última sesión la zona tratada presentaba una piel discretamente atrófica e hipopigmentada. Después de 9 meses de seguimiento la lesión no ha recidivado ni han aparecido otras nuevas.
ComentarioLa elastosis perforante serpiginosa (EPS) es una dermatosis infrecuente que se produce por la eliminación transepidérmica de fibras elásticas engrosadas.
Clásicamente se ha clasificado en tres tipos: idiopática, secundaria al tratamiento con D-penicilamina y asociada con otras enfermedades como síndrome de Down, síndrome de Marfan, osteogénesis imperfecta, síndrome de Ehler-Danlos, acrogeria, morfea, síndrome de Rothmund-Thompson y pseudoxantoma elástico1,2. También han sido descritos algunos casos familiares, con patrones de herencia variable.
Generalmente aparece en la primera etapa de la edad adulta con una proporción entre hombres y mujeres de 4:13. Clínicamente consiste en la aparición de pápulas asintomáticas o levemente pruriginosas, de color de la piel normal o eritematosas, con un tapón de queratina central que sangra al eliminarlo. Estas pápulas tienden a formar un patrón arciforme o serpiginoso. Preferentemente se localizan en la cara, la nuca y las extremidades. En algunas ocasiones las lesiones se resuelven espontáneamente en meses o años, dejando cicatrices hipopigmentadas y atróficas.
Histológicamente se caracteriza por la presencia de fibras elásticas anormales, engrosadas, que se eliminan a través de canales transepidérmicos, que surcan una epidermis acantósica. En ocasiones es posible visualizar un tapón córneo en su superficie, así como células gigantes multinucleadas y un infiltrado inflamatorio pericanalicular4. El estudio mediante tinciones especiales para fibras elásticas (tinción de Verhoeff-Van Gieson u orceína) evidencia la morfología alterada de estas fibras. En los casos inducidos por penicilamina pueden apreciarse ligeras diferencias mediante microscopía óptica y electrónica.
La etiopatogenia de la EPS es desconocida. Se cree que las fibras alteradas serían reconocidas como cuerpo extraño, lo que daría lugar a una reacción granulomatosa con la posterior eliminación de las mismas. La presencia de la reacción granulomatosa alrededor de las fibras elásticas podría tener un papel importante en la fisiopatología de la EPS, así como la de que los queratinocitos epidérmicos que rodean el material elastósico expresan el receptor de la elastina (de 67 kDa), el cual podría estar implicado en su interacción con la elastina y su posterior eliminación5.
El diagnóstico diferencial clínico incluye fundamentalmente al granuloma anular, el prúrigo nodular, la tiña, la sarcoidosis anular, la calcinosis cutánea y la poroqueratosis de Mibelli.
Desde el punto de vista histopatológico con la colagenosis reactiva perforante, genodermatosis infrecuente heredada habitualmente bajo un patrón autosómico dominante y sus variantes adquiridas, asociadas normalmente a disfunción renal o diabetes mellitus: enfermedad de Kyrle y foliculitis perforante.
La mayoría de los tratamientos son insatisfactorios, aunque se han descrito buenos resultados cosméticos con crioterapia6, electrocoagulación, tazaroteno, imiquimod, hidrocortisona subcutánea, tiras de celofán, ácido bicloroacético, láser de CO2 e isotretinoína oral.
En el caso presentado destacamos como inusual la edad tardía de aparición y su presentación como lesión única. Creemos que a pesar de su buena respuesta inicial a la crioterapia puede tratarse del inicio del cuadro y es posible la aparición de nuevas lesiones en el futuro.


