









El descubrimiento de nuevos síndromes autoinflamatorios y nuevas mutaciones está avanzando a una velocidad vertiginosa en los últimos años. La segunda parte de la revisión está centrada en el estudio de los síndromes histiocítico-macrofágicos y de los síndromes vasculopáticos, incluyendo al final del texto una tabla con las alternativas terapéuticas de estos síndromes autoinflamatorios y sus mutaciones genéticas.
The discovery of new autoinflammatory syndromes and novel mutations has advanced at breakneck speed in recent years. Part 2 of this review focuses on vasculitis syndromes and the group of histiocytic and macrophage activation syndromes. We also include a table showing the mutations associated with these autoinflammatory syndromes and treatment alternatives.
El término artritis granulomatosa pediátrica asociada a NOD-2 es una enfermedad hereditaria familiar, autosómica dominante conocida también como síndrome de Blau o sarcoidosis de inicio temprano, que presenta mutaciones en el dominio NACHT del NOD-2 (también conocido como CARD15). Al igual que los NLRP, el NOD-2 es otro receptor intracitoplasmático implicado en la respuesta inmune innata frente a bacterias1, cuyo estímulo ocasiona la formación de lesiones de tipo granulomatoso por activación del NF-kB. Los NLRP y los NOD pueden interactuar y retroalimentarse entre ellos, dando lugar a cuadros clínicos mixtos. Muchos pacientes sin historia familiar presentan mutaciones de novo en NOD2, con formas clínicas incompletas. La expresión fenotípica es muy variable incluso entre los casos familiares y entre pacientes con las mismas mutaciones. La artritis granulomatosa pediátrica presenta un fenotipo similar con una inflamación ocular, articular y cutánea.
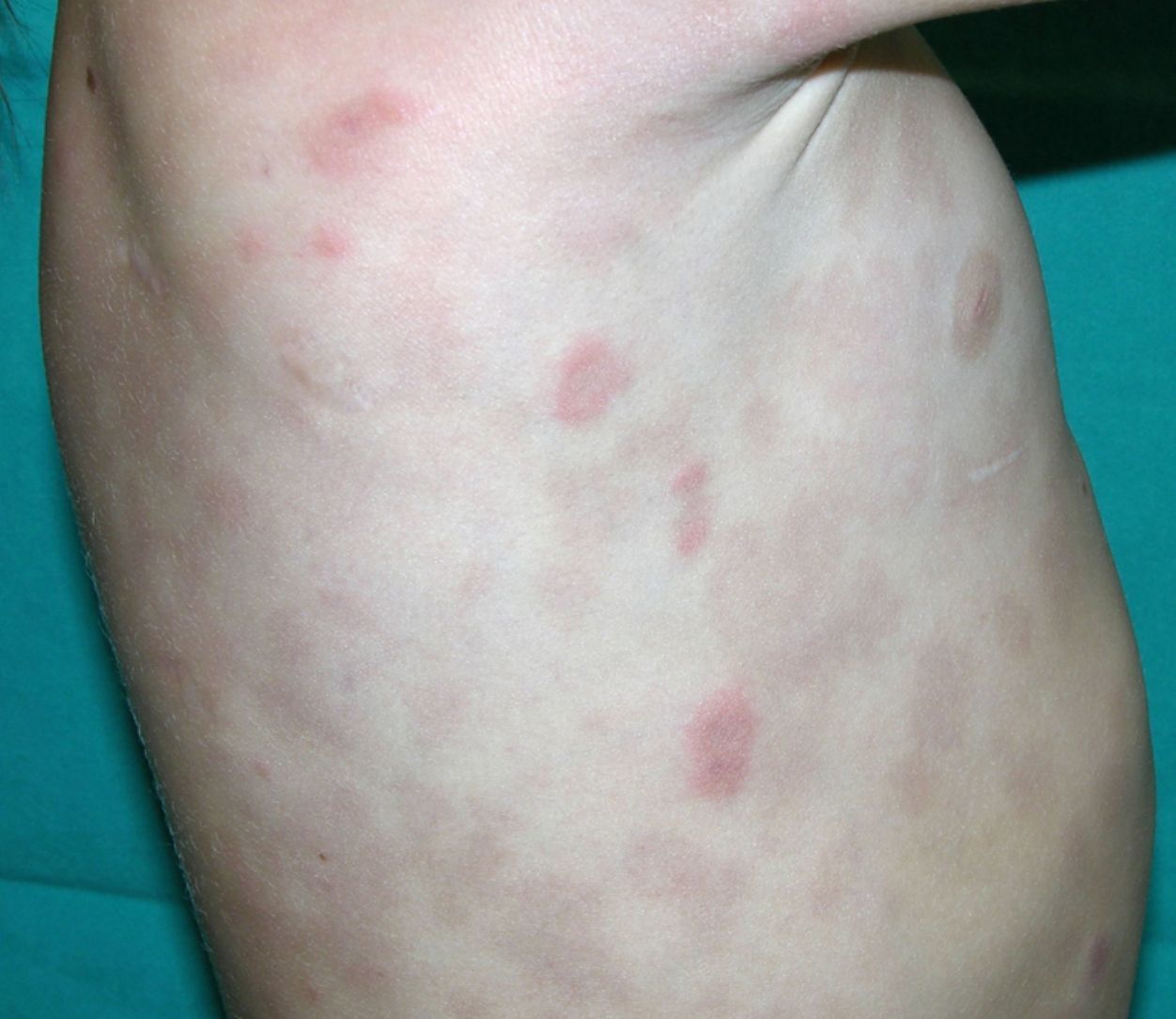
Clínica cutáneaLa afectación cutánea es una de las manifestaciones fundamentales del síndrome de Blau. Desde la primera infancia los pacientes presentan pápulas o papulonódulos asintomáticos, eritematosos o marronáceos (fig. 1), localizados o generalizados que pueden tornarse más o menos liquenoides y afectar las palmas y las plantas. Se han descrito otras manifestaciones cutáneas en las alteraciones del NOD2 como cuadros ictiosiformes2, paniculitis muy similares al eritema nudoso (fig. 2) y vasculitis leucocitoclástica. La forma más frecuente de presentación y que nos debería hacer pensar en esta entidad es una erupción leve de pápulas marronáceas generalizadas de inicio temprano, que a menudo presenta una mejoría espontánea en los primeros años3. La histopatología es similar a la sarcoidosis, con granulomas no caseificantes con histiocitos, linfocitos, eosinófilos (fig. 3) y células gigantes multinucleadas que aparecen intensamente positivas con la tinción de ácido periódico de Schiff. En la microscopia electrónica se pueden apreciar los llamados «cuerpos en coma» en las células epitelioides3.


.")
Es característica la presencia de sinovitis crónica poliarticular, tenosinovitis hipertrófica simétrica y uveítis anterior granulomatosa recurrente, aunque es más habitual la panuveítis. La afectación articular es la más frecuente e importante de las manifestaciones, iniciándose antes de los 10 años en las manos, las muñecas, los tobillos, las rodillas y los codos. Se trata de lesiones quísticas no dolorosas que con el tiempo, de forma lenta y progresiva, adquieren una importante tumefacción, pero sin limitar la movilidad hasta varias décadas después del inicio, apreciándose en la membrana sinovial infiltrados inflamatorios granulomatosos formados por células gigantes multinucleadas. La uveítis anterior se puede complicar con afectación del polo posterior, además de la aparición de cataratas, glaucoma y ceguera, siendo la clínica ocular de presentación bilateral en la mayoría de los casos3. Con menor frecuencia esta infiltración granulomatosa afecta al hígado, a las glándulas salivares, al pulmón, al riñón, al sistema nervioso y a las arterias, generando neuropatía craneal, arteritis de grandes vasos, hipertensión maligna e infartos cerebrales. También se pueden ver implicados los ganglios linfáticos pero, a diferencia de la sarcoidosis4, sin afectación de los hilios y en contraposición a otras enfermedades crónicas autoinflamatorias la amiloidosis secundaria es muy rara5.
Síndrome HEl síndrome H es causado por mutaciones en el gen SLC29A3, localizado en el cromosoma 10q22. Con la letra «H» se engloban varias características asociadas a este síndrome como la hiperpigmentación, la hipertricosis, la hepatoesplenomegalia, el hipogonadismo, las anomalías cardíacas (heart anomalies) y pérdida de audición (hearing loss).
Clínica cutáneaEs muy característica la aparición progresiva de placas hiperpigmentadas e hipertricóticas (fig. 4), con una marcada esclerosis en las extremidades y el abdomen, afectando con mayor frecuencia a la mitad inferior del cuerpo. Se inicia en la mayoría de los pacientes como una hiperpigmentación en la cara interna de los mulsos, que en su progresión suele respetar las rodillas y las nalgas. Algunos pacientes presentan además una descamación ictiosiforme. La biopsia cutánea muestra una hiperpigmentación de la capa basal con acantosis e hiperplasia similar a la queratosis seborreica, marcada fibrosis de la dermis y el tejido celular subcutáneo, que es leve en la dermis papilar y mucho más intensa en el subcutis, con obliteración progresiva del tejido graso (fig. 5). Se acompaña de un infiltrado compuesto principalmente por numerosos histiocitos pequeños de citoplasma claro6 (CD68+, S100+, CD1a–), observándose en ocasiones imágenes de emperipolesis. Con microscopia electrónica es característica la visualización de un retículo endoplásmico distendido y escasez de lisosomas.

.")
Se han descrito asociadas a este síndrome la presencia de sordera, baja estatura, contracturas en flexión, ginecomastia, venas dilatadas en las piernas y sobre la pared abdominal, anomalías cardíacas, cardiopatía isquémica precoz7, hepatoesplenomegalia, masas escrotales, hipogonadismo, retraso de la pubertad, azoospermia e hiperglucemia/diabetes mellitus8.
Dermatosis neutrofílica crónica atípica con lipodistrofia y temperatura elevadaEl síndrome de dermatosis neutrofílica crónica atípica con lipodistrofia y temperatura elevada (CANDLE) es el paradigma de enfermedad asociada a disfunción del proteasoma-inmunoproteasoma9. Estos son complejas estructuras proteicas intracelulares que escinden y eliminan residuos proteicos marcados por la ubiquitina que ya no son de utilidad. El proteasoma está presente de forma constitutiva en la célula, pero cuando las necesidades catalíticas se ven aumentadas (ante una infección o estímulo físico) es necesario formar un complejo inducible denominado inmunoproteasoma. Si hay defectos en la formación del inmunoproteasoma o este es hipofuncionante se produce un importante cúmulo de proteínas marcadas por ubiquitina en el interior de los macrófagos, generando un importante estrés celular e incremento de la producción de interferón tipo 1 debido a la activación secundaria de la vía de señalización de las JAK-quinasas, cerrándose así un círculo vicioso abocado a la continua formación y acumulación de desechos proteicos intracelulares. Las características principales del CANDLE incluyen episodios de fiebre recurrente, inflamación visceral, lipodistrofia y lesiones cutáneas fijas sin tendencia a la autorresolución, que se manifiestan en el primer año de vida, generalmente durante las primeras semanas. Los estudios genéticos revelan mutaciones en varios de los genes que codifican diferentes subunidades del proteasoma/inmunoproteasoma, como la β5i, β1i, β7 o α3 que condicionan la disminución de la actividad catalítica del inmunoproteasoma o impiden su ensamblaje. Aunque en los pacientes con síndrome de CANDLE lo más frecuente es encontrar mutaciones bialélicas en el gen PSMB8, que codifica la subunidad β5i, se han descrito recientemente herencias digénicas monoalélicas que codifican diferentes subunidades10 y mutaciones en POMP en un paciente (tabla 1).
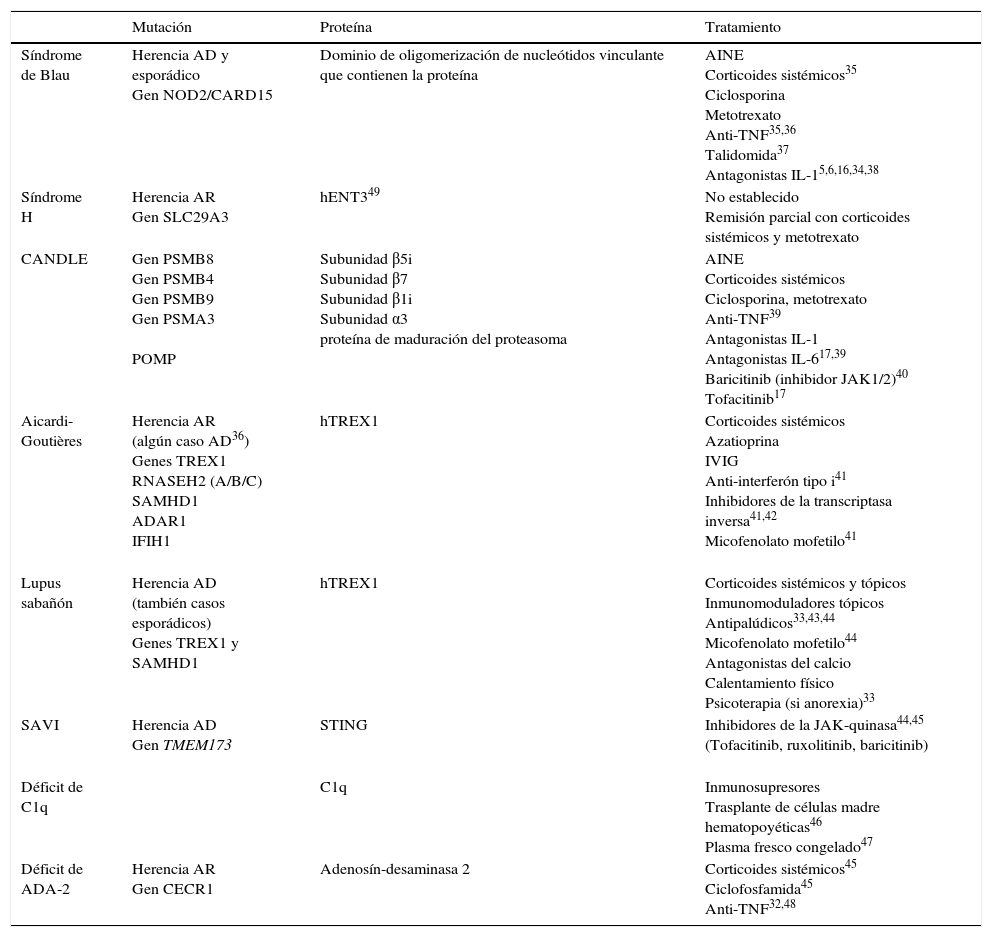
Mutaciones genéticas y alternativas terapéuticas
| Mutación | Proteína | Tratamiento | |
|---|---|---|---|
| Síndrome de Blau | Herencia AD y esporádico Gen NOD2/CARD15 | Dominio de oligomerización de nucleótidos vinculante que contienen la proteína | AINE Corticoides sistémicos35 Ciclosporina Metotrexato Anti-TNF35,36 Talidomida37 Antagonistas IL-15,6,16,34,38 |
| Síndrome H | Herencia AR Gen SLC29A3 | hENT349 | No establecido Remisión parcial con corticoides sistémicos y metotrexato |
| CANDLE | Gen PSMB8 Gen PSMB4 Gen PSMB9 Gen PSMA3 POMP | Subunidad β5i Subunidad β7 Subunidad β1i Subunidad α3 proteína de maduración del proteasoma | AINE Corticoides sistémicos Ciclosporina, metotrexato Anti-TNF39 Antagonistas IL-1 Antagonistas IL-617,39 Baricitinib (inhibidor JAK1/2)40 Tofacitinib17 |
| Aicardi-Goutières | Herencia AR (algún caso AD36) Genes TREX1 RNASEH2 (A/B/C) SAMHD1 ADAR1 IFIH1 | hTREX1 | Corticoides sistémicos Azatioprina IVIG Anti-interferón tipo i41 Inhibidores de la transcriptasa inversa41,42 Micofenolato mofetilo41 |
| Lupus sabañón | Herencia AD (también casos esporádicos) Genes TREX1 y SAMHD1 | hTREX1 | Corticoides sistémicos y tópicos Inmunomoduladores tópicos Antipalúdicos33,43,44 Micofenolato mofetilo44 Antagonistas del calcio Calentamiento físico Psicoterapia (si anorexia)33 |
| SAVI | Herencia AD Gen TMEM173 | STING | Inhibidores de la JAK-quinasa44,45 (Tofacitinib, ruxolitinib, baricitinib) |
| Déficit de C1q | C1q | Inmunosupresores Trasplante de células madre hematopoyéticas46 Plasma fresco congelado47 | |
| Déficit de ADA-2 | Herencia AR Gen CECR1 | Adenosín-desaminasa 2 | Corticoides sistémicos45 Ciclofosfamida45 Anti-TNF32,48 |
Las manifestaciones cutáneas de CANDLE consisten en placas edematosas eritematovioláceas anulares de bordes sobrelevados, y aunque afectan principalmente al tronco (fig. 6), también se ven comprometidas la zona peribucal, la periocular donde presentan un eritema violáceo persistente11 y las extremidades, fundamentalmente las articulaciones interfalángicas. Estas lesiones curan espontáneamente dejando hiperpigmentación o máculas equimóticas residuales, pero recurren con la frecuencia suficiente como para considerar una afectación cutánea crónica y persistente en estos pacientes. El estudio anatomopatológico muestra densos infiltrados inflamatorios de localización dérmica, perivascular e intersticiales, posible afectación del tejido celular subcutáneo con menor frecuencia, ausencia de vasculitis y presencia no constante de cariorrexis. Se compone de células mieloides mononucleares atípicas de núcleos grandes, abigarrados, alargados o arriñonados y citoplasma eosinófilo escaso11, acompañado de un variable número de neutrófilos y eosinófilos (fig. 7). Este infiltrado celular muestra una inmunohistoquímica positiva para mieloperoxidasa, lisozima, Mac387, CD68 (KP1) y CD16312,13 (fig. 8), revelando una doble población de células mieloides y macrófagos. Además, la presencia de células dendríticas plasmocitoides CD123, potentes productoras de interferón tipo i, indica que esta citoquina tiene una importancia más que significativa en el síndrome CANDLE.

.")
.")
Con menor frecuencia los pacientes pueden presentar nódulos violáceos que dejan una hiperpigmentación residual, hipertricosis, acantosis nigricans y alopecia areata14, siendo muy característico y definitorio del síndrome la aparición de una lipodistrofia progresiva en la cara, las extremidades superiores y el tronco.
Clínica asociadaSegún lo descrito en la literatura, siempre están presentes los episodios de fiebre elevada (superior a 38°C) recurrentes e incluso diarios, el bajo peso y la baja talla15. Pueden asociar adenopatías, hepatoesplenomegalia y artralgia sin artritis, además de otros muchos eventos inflamatorios como condritis, conjuntivitis, epiescleritis nodular, epididimitis, nefritis, otitis, parotiditis o meningitis aséptica. Además los pacientes con CANDLE asocian algunos rasgos dismórficos15 como puente nasal ancho, labios gruesos o eversión del labio inferior. En el análisis de sangre es característica la elevación de reactantes de fase aguda, anemia crónica, aumento de las enzimas hepáticas y linfopenia transitoria durante brotes de la enfermedad.
Síndromes vasculopáticosDentro de este grupo incluiremos las interferonopatías y los cuadros causados por el déficit de adenosindeaminasa-2 (ADA-2).
Interferonopatías tipo 1Los interferones (IFN) tipo 1 son un grupo de citoquinas fundamentales en la conexión entre la inmunidad innata y la adquirida. El ADN viral, reconocido por su receptor (IFNAR), activa la señalización dependiente de STING (estimulador de genes de interferón) produciendo IFN y otras citoquinas proinflamatorias16. El aumento de IFN estimula el inmunoproteasoma que activa con mayor frecuencia la vía JAK-STAT y se genera más cantidad de IFN, intensificando y perpetuando la respuesta inmune y los cuadros de autoinflamación. A continuación vamos a describir las interferonopatías más relevantes en dermatología con su cuadro clínico característico.
Síndrome de Aicardi-GoutièresEl síndrome de Aicardi-Goutières es una enfermedad neurodegenerativa genéticamente muy heterogénea, con un alto grado de consanguinidad, caracterizada por la afectación cerebral y cutánea. En la mayoría de los pacientes es transmitida con herencia autosómica recesiva, aunque se han descrito casos de transmisión autosómica dominante. Se han identificado las mutaciones responsables de la enfermedad en 7 genes: TREX1, que codifica una exonucleasa 3’-5’, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, IFIH1 y ADAR117. Las mutaciones asociadas causan una deficiente destrucción de material genético viral, necesaria para regular a la baja la producción de interferón en las células infectadas. Esta disregulación del IFN-alfa genera situaciones proinflamatorias que desencadenan cuadros vasculíticos secundarios, como la encefalopatía o las lesiones de acrocianosis. Aunque puede ser congénito, es más común que se desarrolle en el primer año de vida en niños nacidos a término sanos.
Clínica cutáneaDe forma característica los pacientes presentan lesiones de tipo sabañón, fundamentalmente en los dedos de las manos, aunque también pueden afectarse los pies y los pabellones auriculares, más frecuentes en invierno y con necrosis acral asociada. Además de las manifestaciones cutáneas asociadas más conocidas, como la fotosensibilidad, la acrocianosis, la púrpura petequial en la mitad superior del tronco y la cara, la edematización de manos y pies, el eritema periungueal, la distrofia ungueal o los pies fríos, se han descrito casos de erupción tipo blueberry muffin baby imitando el fenotipo clínico de una infección viral congénita tipo TORCH, a modo de maculopápulas rojo-azuladas generalizadas18. En esta última situación el estudio histopatológico es compatible con una hematopoyesis extramedular, apreciándose un infiltrado polimorfo localizado en la dermis y en el tejido celular subcutáneo, y compuesto predominantemente de células precursoras eritroides y mieloides, con inmunotinción positiva para glicoforina-A, mieloperoxidasa, CD20 y CD3 confirmando así la presencia de eritrocitos, leucocitos, células B y células T respectivamente18.
Clínica asociadaLos pacientes desarrollan una encefalopatía subaguda grave progresiva hasta su estabilización, caracterizada por la asociación de calcificación de los ganglios basales, leucodistrofia, microcefalia evolutiva y disfunción piramidal-extrapiramidal que se acompaña de epilepsia y cuadros febriles asépticos. Se han descrito casos de artropatía crónica con contracturas progresivas acrales y ulceración de la mucosa oral19. En los análisis destacan niveles elevados de IFN-alfa, neopterina, biopterina20 y linfocitosis que también se observa en el líquido cefalorraquídeo, considerando la elevación del IFN-alfa en líquido cefalorraquídeo un criterio mayor de diagnóstico21. Además estos pacientes pueden presentar trombocitopenia, aumento de las enzimas hepáticas y en algunos casos hiperpoligammaglobulinemia y anemia hemolítica Coombs positiva. Todas estas asociaciones tienen una expresión clínica muy variable en cada paciente, y ninguna es necesaria para el diagnóstico19.
Lupus sabañón familiarEste síndrome es una rara forma familiar de lupus eritematoso cutáneo inducido por el frío, que puede ser esporádico o con menor frecuencia hereditario, generalmente causado por una mutación en TREX1. TREX1 es la principal exonucleasa de reparación intracelular cuya deficiencia conduce a la acumulación de ácidos nucleicos, con una activación de la inmunidad innata acompañada de aumento de INF que favorece el desarrollo de la autoinmunidad6.
Clínica cutáneaDesde la infancia estos pacientes desarrollan placas violáceas de aspecto perniótico y de localización acral que pueden ulcerarse, muy similares tanto clínica como histopatológicamente a las observadas en el síndrome de Aicardi-Goutières. La histología revela una dermatitis de interfase con degeneración vacuolar en la capa basal, acompañada de un denso infiltrado inflamatorio linfohistiocitario de localización perivascular en la dermis, junto a la presencia de mucina en la dermis reticular. La IFD revela un depósito basal de IgM, IgA y C3 con depósitos perivasculares de C3 y fibrinógeno22,23.
Clínica asociadaLos pacientes presentan ocasionalmente afectación sistémica con artralgias o artritis no erosiva. En el análisis sanguíneo pueden aparecer pancitopenia leve, hipergammaglobulinemia, FR, ANA y anti-Ro/SSA23, siendo las crioglobulinas y crioaglutininas negativas. Hasta el 20% de los pacientes desarrollan lupus eritematoso sistémico24 y hay discrepancia de su relación con la anorexia.
Vasculopatía asociada a proteína estimuladora de genes de interferón con inicio en la infanciaComo hemos explicado anteriormente, el ARN y ADN viral constituyen la principal causa biológica reconocida para la activación de la proteína STING (estimulador de genes de interferón), una proteína transmembrana que se localiza en el retículo endoplásmico y que induce la producción de interferón. La vasculopatía asociada a STING con inicio en la infancia se debe a mutaciones hiperfuncionantes en STING25,26, proteína que se expresa ampliamente en las células endoteliales y las células alveolares. La hiperfunción de STING ocasiona inflamación en las células endoteliales generando fenómenos de vasooclusión, así como lesiones pulmonares, posiblemente debido a la activación de macrófagos o neumocitos alveolares. La liberación de IFN conduce a una retroalimentación positiva, ya que desencadena directamente la transcripción de otras citoquinas proinflamatorias y activa las JAK-quinasas, que fosforilan y dimerizan STAT1/STAT2, que a su vez inician la transcripción de genes de interferón.
Clínica cutáneaEl cuadro clínico de estos pacientes se inicia en los primeros meses de vida como una erupción telangiectásica, pustulosa o ampollar (o una combinación) localizada en zonas acrales y mejillas con posterior progresión, inducida sobre todo por la exposición al frío, hacia nódulos y placas violáceas acrales que en la mayoría de los pacientes se necrosan, pudiendo afectar amplias zonas con pérdida importante de tejido. Son características la tortuosidad capilar del lecho ungueal, las telangiectasias en la nariz, las mejillas, las extremidades y el paladar duro, pudiendo observarse en algunos pacientes perforación del tabique nasal, uñas distróficas, resorción de falanges distales y gangrenas digitales que requieren la amputación26. En la histopatología se aprecia una densa reacción inflamatoria localizada en la dermis de predominio neutrofílico con vasculitis leucocitoclástica y microangiopatía trombótica en los pequeños vasos dérmicos26, en algunos hay depósitos de IgM, C3 y fibrina.
Clínica asociadaLos pacientes con este síndrome tienen episodios recurrentes de fiebre no muy elevada y pueden presentar retraso del crecimiento, importante enfermedad pulmonar con adenopatías hiliares o paratraqueales, fibrosis intersticial y cambios enfisematosos producidos por un infiltrado inflamatorio linfocítico27. En el análisis de sangre destaca un aumento de VSG y PCR y ocasional presencia de ANA, c-ANCA y anticuerpos anti-fosfolípidos, que suelen normalizarse con el tiempo.
Déficit de C1qLa proteína C1q es una proteína que forma parte del complejo C1 del sistema del complemento. Además de sus funciones en la activación de la cascada del complemento, el C1q inhibe la producción de interferón por las células dendríticas plasmocitoides y las células mononucleares de sangre periférica. Las mutaciones con pérdida de función de C1q causan un fenotipo similar al lupus eritematoso sistémico (LES) en un 90% de los casos28.
Clínica cutáneaLa clínica cutánea del déficit de C1q incluye un eritema en alas de mariposa en la cara, lesiones tipo lupus discoide y aparición de fenómeno de Raynaud similar al detectado en el LES29.
Clínica asociadaLas manifestaciones extracutáneas de este déficit de C1q incluyen la glomerulonefritis segmental mesangial, que es diferente a la nefritis típica del LES, síntomas neurológicos e infecciones recurrentes graves que se deben al déficit de la función de la vía clásica del complemento. En el análisis sanguíneo los niveles de CH50 y C1q en plasma son prácticamente de cero, sin embargo, los niveles de C2, C4 y C1 inhibidor son normales o elevados con positividad de los ANA, el FR y los anticuerpos anti-Sm. Las biopsias de las lesiones muestran vacuolización de la membrana basal con presencia de depósitos de IgG, IgM, IgA y C3 en la IFD. La asociación de una clínica similar al LES junto a una clínica de inmunodeficiencia es altamente sugestiva de la deficiencia de factores del complemento.
Déficit de adenosín-deaminasa 2El déficit de adenosín deaminasa-2 se relaciona con cuadros clínicamente inespecíficos superponibles a una poliarteritis nodosa con presencia de una vasculopatía necrosante de mediano y pequeño vaso asociada a lívedo reticular, con una marcada predisposición familiar, por lo que también se le ha denominado poliarteristis nodosa familiar. Niveles bajos en sangre de deanosín deaminasa-2 se han relacionado con alteraciones endoteliales y con una mayor activación de los macrófagos, situaciones que probablemente causan vasculitis y accidentes cerebrovasculares en estos pacientes30,31. La clínica es muy variable en cuanto a la edad de inicio, la gravedad y la participación de órganos, incluso dentro de las familias que presentan las mismas mutaciones32.
Clínica cutáneaLa clínica cutánea típica de este síndrome incluye la presencia de alteraciones relacionadas con un mal flujo arterial, entre las que se incluyen la livedo reticular, el fenómeno de Raynaud, la isquemia y la necrosis digital. La biopsia de las lesiones muestra periarteritis, necrosis fibrinoide y destrucción de la lámina elástica de las arteriolas cutáneas. También se pueden ver vasculitis leucocitoclástica y paniculitis.
Clínica asociadaAdemás de la clínica cutánea estos pacientes presentan episodios febriles, aneurismas frecuentes en las arterias renales, mesentéricas o coronarias. Secundariamente a la isquemia pueden verse manifestaciones renales, gastrointestinales, genitales, musculoesqueléticas, cardiovasculares y neurológicas. Son de especial relevancia la hipertensión arterial de origen renal y alto riesgo de ictus cerebral de forma precoz en la edad infantil31.
Manejo de los síndromes autoinflamatoriosEl conocimiento de las mutaciones genéticas asociadas a los síndromes que hemos descrito en esta revisión (tabla 2) nos está permitiendo identificar y mejorar la calidad de vida de estos pacientes. El manejo de estas enfermedades está muy relacionado con el control de la respuesta inmunológica que las ocasiona, es por esto que muchos de estos síndromes tienen un tratamiento específico o se benefician del bloqueo de determinadas moléculas proinflamatorias (tabla 1). Se han utilizado los anti-TNF y los antagonistas de la IL-1. El perfil de éxito y la seguridad de los antagonistas de la IL-1, como anakinra (antagonista del receptor de IL-1), rilonacept (proteína de fusión dimérica) o canakinumab (anticuerpo monoclonal totalmente humanizado dirigido específicamente contra la IL-1β) en el tratamiento de CAPS y DIRA han animado a ampliar su uso en otros síndromes autoinflamatorios, como los síndromes de fiebres periódicas y otras enfermedades con disregulación inmunitaria, como la enfermedad de Still, la de Behcet o la de Schnitzler. Además, el hecho de que la acumulación de sustratos metabólicos, tales como urato monosódico, colesterol o glucosa pueden estimular el inflamasoma NLRP3 por estrés metabólico y el papel secundario de la IL-1, podría justificar el empleo de los anti IL-1 en enfermedades tan prevalentes como la gota, la diabetes mellitus o la enfermedad coronaria33,34. En el síndrome CANDLE el tratamiento con baricitinib (inhibidor de la JAK 1/2) está consiguiendo resultados alentadores.
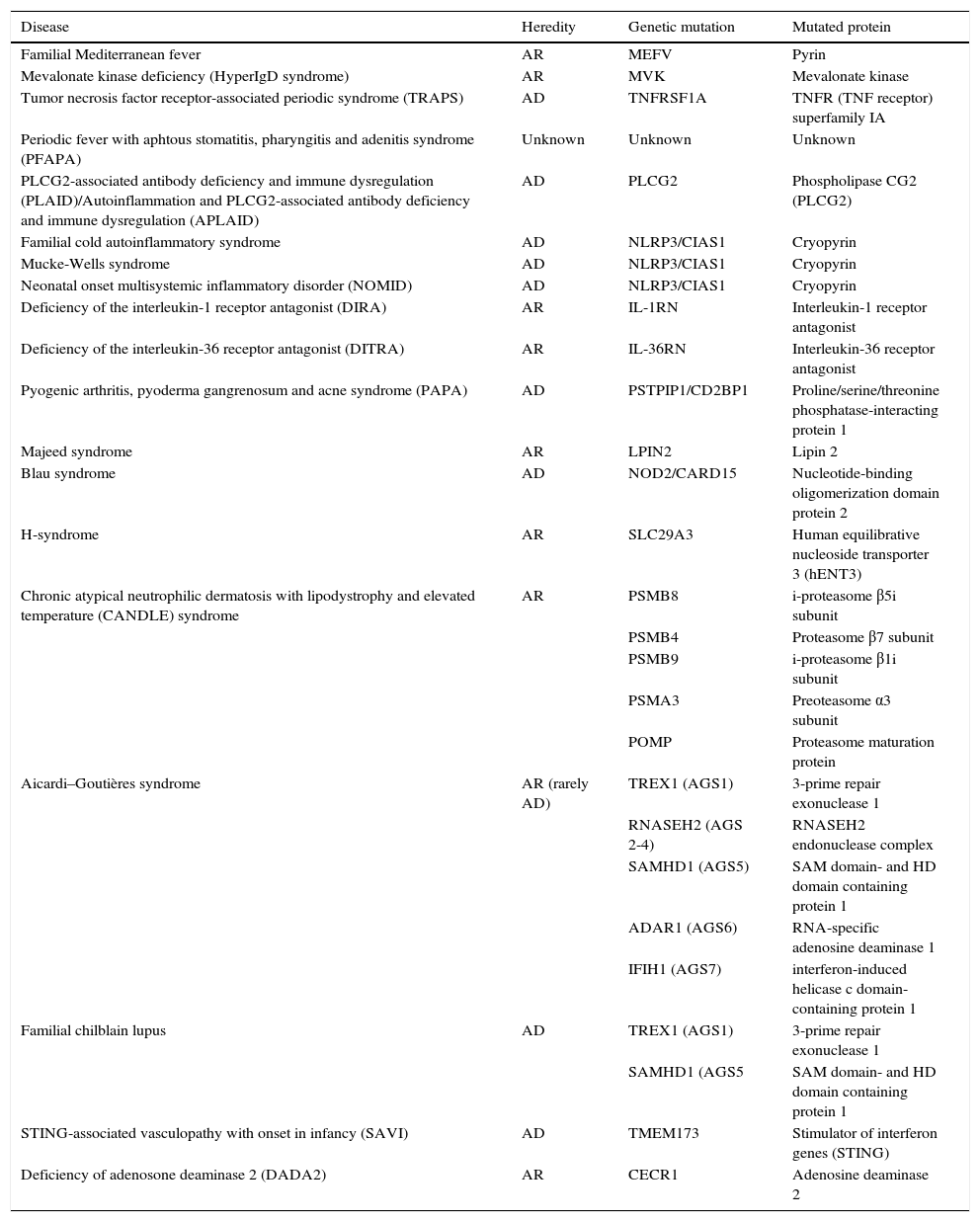
Resumen de las principales enfermedades autoinflamatorias y sus mutaciones
| Disease | Heredity | Genetic mutation | Mutated protein |
|---|---|---|---|
| Familial Mediterranean fever | AR | MEFV | Pyrin |
| Mevalonate kinase deficiency (HyperIgD syndrome) | AR | MVK | Mevalonate kinase |
| Tumor necrosis factor receptor-associated periodic syndrome (TRAPS) | AD | TNFRSF1A | TNFR (TNF receptor) superfamily IA |
| Periodic fever with aphtous stomatitis, pharyngitis and adenitis syndrome (PFAPA) | Unknown | Unknown | Unknown |
| PLCG2-associated antibody deficiency and immune dysregulation (PLAID)/Autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation (APLAID) | AD | PLCG2 | Phospholipase CG2 (PLCG2) |
| Familial cold autoinflammatory syndrome | AD | NLRP3/CIAS1 | Cryopyrin |
| Mucke-Wells syndrome | AD | NLRP3/CIAS1 | Cryopyrin |
| Neonatal onset multisystemic inflammatory disorder (NOMID) | AD | NLRP3/CIAS1 | Cryopyrin |
| Deficiency of the interleukin-1 receptor antagonist (DIRA) | AR | IL-1RN | Interleukin-1 receptor antagonist |
| Deficiency of the interleukin-36 receptor antagonist (DITRA) | AR | IL-36RN | Interleukin-36 receptor antagonist |
| Pyogenic arthritis, pyoderma gangrenosum and acne syndrome (PAPA) | AD | PSTPIP1/CD2BP1 | Proline/serine/threonine phosphatase-interacting protein 1 |
| Majeed syndrome | AR | LPIN2 | Lipin 2 |
| Blau syndrome | AD | NOD2/CARD15 | Nucleotide-binding oligomerization domain protein 2 |
| H-syndrome | AR | SLC29A3 | Human equilibrative nucleoside transporter 3 (hENT3) |
| Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome | AR | PSMB8 | i-proteasome β5i subunit |
| PSMB4 | Proteasome β7 subunit | ||
| PSMB9 | i-proteasome β1i subunit | ||
| PSMA3 | Preoteasome α3 subunit | ||
| POMP | Proteasome maturation protein | ||
| Aicardi–Goutières syndrome | AR (rarely AD) | TREX1 (AGS1) | 3-prime repair exonuclease 1 |
| RNASEH2 (AGS 2-4) | RNASEH2 endonuclease complex | ||
| SAMHD1 (AGS5) | SAM domain- and HD domain containing protein 1 | ||
| ADAR1 (AGS6) | RNA-specific adenosine deaminase 1 | ||
| IFIH1 (AGS7) | interferon-induced helicase c domain-containing protein 1 | ||
| Familial chilblain lupus | AD | TREX1 (AGS1) | 3-prime repair exonuclease 1 |
| SAMHD1 (AGS5 | SAM domain- and HD domain containing protein 1 | ||
| STING-associated vasculopathy with onset in infancy (SAVI) | AD | TMEM173 | Stimulator of interferon genes (STING) |
| Deficiency of adenosone deaminase 2 (DADA2) | AR | CECR1 | Adenosine deaminase 2 |
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses


