




Las enfermedades monogénicas autoinflamatorias son un grupo de enfermedades emergentes y heterogéneas en continuo estudio y desarrollo en la actualidad. Nuestro objetivo es revisar estas enfermedades desde el punto de vista de su etiopatogenia y principales manifestaciones, con el fin de proponer una clasificación, basada en las características clinicopatológicas de las lesiones cutáneas típicas, que resulte de utilidad en la práctica clínica habitual de los dermatólogos. El texto está enfocado en el diagnóstico de estos síndromes durante la edad pediátrica, ya que es el periodo habitual de aparición de los primeros síntomas y signos. La primera parte de la revisión se centrará en el desarrollo de los síndromes urticariformes, que incluyen a su vez las criopirinopatías y los síndromes hereditarios asociados a fiebres periódicas, y de los síndromes pustulosos, resumiendo al final del texto las alternativas terapéuticas de estos síndromes autoinflamatorios y sus mutaciones genéticas.
Monogenic autoinflammatory diseases are a heterogeneous emergent group of conditions that are currently under intensive study. We review the etiopathogenesis of these syndromes and their principal manifestations. Our aim is to propose a classification system based on the clinicopathologic features of typical skin lesions for routine clinical use in dermatology. Our focus is on diagnosis in pediatric practice given that this is the period when the signs and symptoms of these syndromes first appear. In Part 1 we discuss the course of urticaria-like syndromes, which include cryopyrin-associated periodic conditions and hereditary periodic fever syndromes. Pustular syndromes are also covered in this part. Finally, we review the range of therapies available as well as the genetic mutations associated with these autoinflammatory diseases.
Las enfermedades autoinflamatorias son un grupo de enfermedades en las que se afecta principalmente la inmunidad innata con una respuesta inflamatoria exagerada no dependiente de antígeno. Esto es diferente a lo que ocurre en las enfermedades autoinmunes, donde la alteración de la inmunidad adquirida desempeña un papel clave mediante un incremento de las respuestas dependientes de antígeno. La inmunidad innata constituye la primera línea de defensa contra patógenos y estímulos nocivos, mediante el reconocimiento de marcadores moleculares asociados a patógenos infecciosos (PAMP) o a daño (DAMP), con la posterior activación de múltiples cascadas de señalización inflamatorias y de las principales células efectoras de la inmunidad innata: macrófagos, neutrófilos, mastocitos y células asesinas naturales (NK)1. Aunque cada síndrome tiene manifestaciones clínicas que lo caracterizan, casi todas comparten generalidades como el inicio precoz, en la infancia o incluso en el periodo neonatal, la aparición de brotes recurrentes de fiebre, inflamación multisistémica y la presencia de una gran variedad de manifestaciones cutáneas. Existen en la literatura diversas clasificaciones de las enfermedades autoinflamatorias hereditarias, pero para la presente revisión las orientaremos según su contexto clinicopatológico dermatológico para facilitar su reconocimiento en función de las manifestaciones cutáneas.
Síndromes urticariformesEn este primer apartado hemos incluido 2 grupos de enfermedades, las criopirinopatías y los síndromes de fiebre periódica, que se manifiestan en muchos casos con cuadros urticariformes y lesiones de eritema y edema en placas.
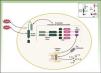
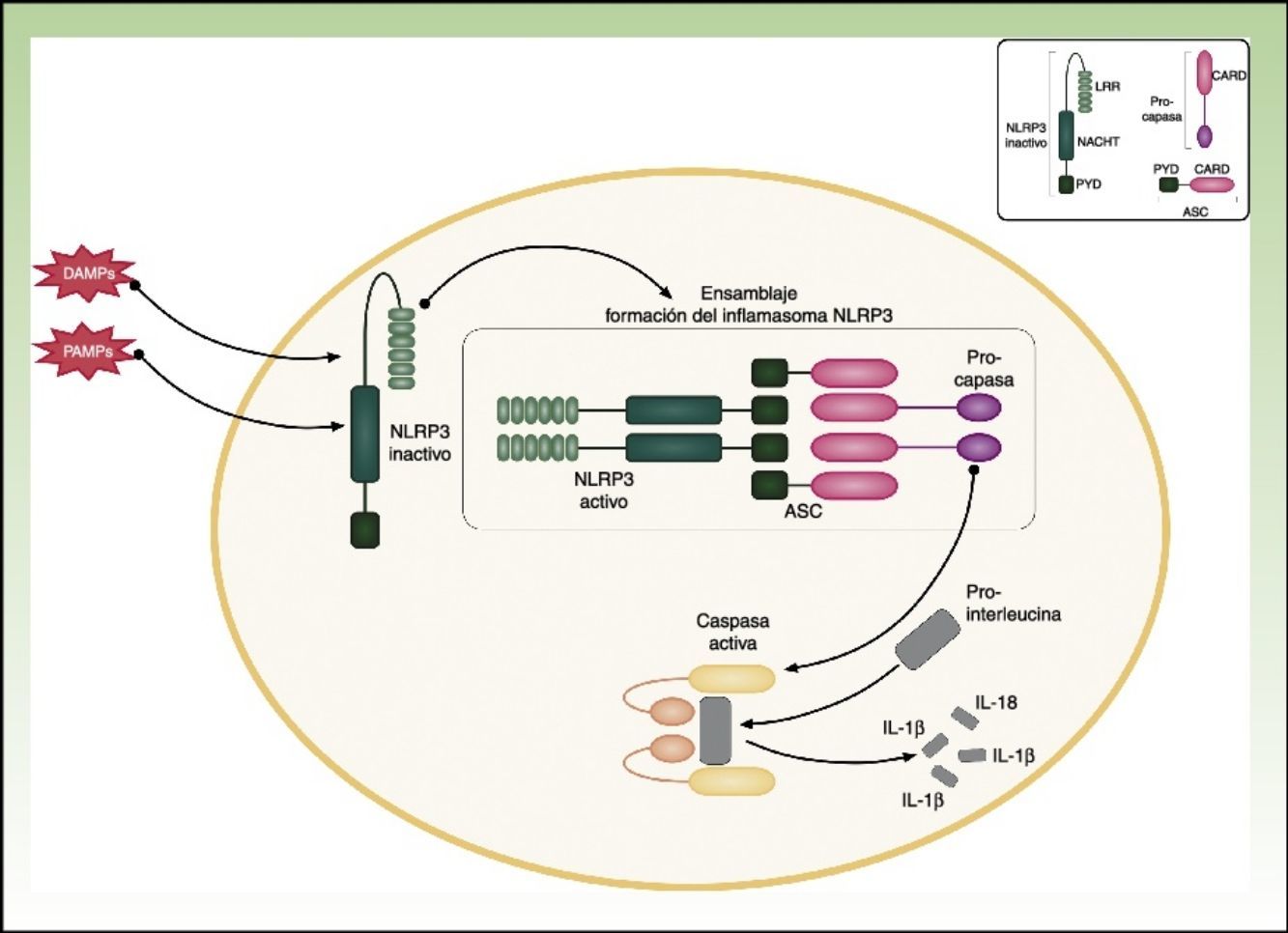
CriopirinopatíasLas criopirinopatías o cryopyrin-associated periodic syndromes (CAPS) engloban un grupo de 3 enfermedades autoinflamatorias alélicas que son el síndrome autoinflamatorio familiar por frío o FCAS, el síndrome de Muckle Wells y el síndrome CINCA o NOMID que corresponde a los acrónimos de Chronic Infantile Neurological Cutaneous Articular Syndrome o Neonatal-Onset Multisystem Inflammatory Disease. Desde el punto de vista genético estas enfermedades se heredan de forma autosómica dominante, tienen una penetrancia variable y comparten una etiopatogenia común, ya que las 3 se deben a mutaciones hiperfuncionantes en el gen NLRP3 (también llamado CIAS1). Este gen codifica la criopirina (NALP3 o PYPAF1), que es una proteína fundamental en la constitución del inflamasoma NLRP3. El inflamasoma es un complejo multiproteico que en respuesta a multitud de estímulos activa a través de la caspasa 1 una serie de interacciones intracelulares que finalizan con la producción de potentes citoquinas proinflamatorias, como la IL-1β y IL-18 (fig. 1). La activación de la criopirina se inicia por el reconocimiento de PAMP o DAMP. Las mutaciones descritas en el gen NLRP3 como causantes de estos síndromes son mutaciones hiperfuncionantes que activan este inflamasoma de forma constitutiva, aumentando la producción de citoquinas proinflamatorias (tabla 1).
Mutaciones genéticas y alternativas terapéuticas
| Mutación | Proteína | Tratamiento | |
|---|---|---|---|
| FMF | Herencia AR gen MEFV | Pirina | Elección: colchicina19 |
| Antagonistas de la IL-16,19,20 | |||
| (anakinra, rilonacept, canakinumab) | |||
| Interferón-alfa, talidomida | |||
| Anti-TNF7,19 | |||
| HIDS | Herencia AR gen MVK (mevalonato quinasa) | Mevalonato quinasa | Corticoides sistémicos19 |
| Colchicina | |||
| AINE21 | |||
| Simvastatina22,23 | |||
| Talidomida24 | |||
| Antagonistas de la IL-16,19,20,25,26 | |||
| Anti-TNF19,27,28 | |||
| TRAPS | Herencia AD gen TNFRSF1A | Receptor de TNF p55 (TNFR1) | Corticoides sistémicos19,29 |
| (no respuesta a colchicina) | |||
| Anti-TNF19,29,30–33Antagonistas IL-17,19,20,25,29,34 | |||
| Antagonistas IL-6 (tocilizumab)29,35,36 | |||
| Tacrólimus37 | |||
| PFAPA | Desconocido | Desconocido | Corticoides sistémicos19,38 |
| Cimetidina | |||
| Colchicina39 | |||
| Amigdalectomía40 | |||
| Antagonistas de la IL-125 | |||
| PLAID/APLAID | Herencia AD gen PLCG2 | fosfolipasa Cg2 (PLCg2) | Altas dosis corticoides sistémicos |
| Antagonistas IL-1 (respuesta parcial)8,9 | |||
| FCAS | Gen NLRP3/CIAS1 AD | Criopirina (NALP3 o PYPAF1) | No exposición al frío Antagonistas IL-17,19,20 |
| MWS | Gen NLRP3/CIAS1 AD | Criopirina (NALP3 o PYPAF1) | Antagonistas IL-16,7,19,20,41 |
| NOMID/CINCA | Gen NLRP3/CIAS1 AD algún caso esporádico7 | Criopirina (NALP3 o PYPAF1) | Antagonistas IL-13,6,7,19,20,42 |
| DIRA | Herencia AR gen IL-1RN | Antagonista del receptor de la interleucina-1 | Antagonistas IL-111,19,20,25 |
| DITRA | Herencia AR casos esporádicos gen IL-36RN | Antagonista del receptor de la interleucina-36 | No establecido7 |
| Acitretino7 | |||
| Corticoides tópicos y sistémicos | |||
| Metotrexato | |||
| Ciclosporina | |||
| Anti-TNF | |||
| Aféresis de granulocitos y monocitos43 | |||
| Antagonistas IL-144 | |||
| PAPA | Gen PSTPIP1/CD2BP1 AD | PSTPIP1/CD2BP1 | Corticoides sistémicos7,19 |
| Ciclosporina7,talidomida7, dapsona, tacrolimus | |||
| IVIG7 | |||
| Antagonistas IL-119,25 | |||
| Combinación de isotretinoína y anakinra19 | |||
| Anti-TNF14,15,19 (quizás de elección) | |||
| Síndrome de Majeed | Herencia AR gen LPIN2 | LPIN2 fosfatasa fosfatidato | AINE |
| Corticoides, interferón gamma, bifosfonatos | |||
| Anti-TNF | |||
| Antagonistas IL-17,16,17,25 | |||
| PAAND | Herencia AD gen MEFV | Pirina | Antagonistas IL-118 (un paciente con buena respuesta) |
El FCAS o urticaria familiar por frío es el síndrome de menor agresividad de todas las criopirinopatías. Las manifestaciones clínicas se inician en el primer año de vida, y a menudo son perceptibles ya desde el periodo neonatal, siendo característica la existencia de un lapso de entre 2 y 7horas desde la exposición al frío y la aparición de la clínica, la cual suele desaparecer tras aproximadamente unas 12horas2. Las lesiones no son provocadas por el contacto con objetos fríos.
Clínica cutáneaLos pacientes suelen presentar pápulas y placas eritematoedematosas, similares a una urticaria aguda común, pero con mayor simetría de las lesiones. En la biopsia cutánea destaca la presencia de un infiltrado dérmico de predominio mayoritariamente neutrofílico y de disposición perivascular, a diferencia de lo que se observa en las urticarias comunes idiopáticas, donde predomina un infiltrado linfocitario y eosinofílico sin vasculitis. No obstante, el grado de edema en la dermis y el predominio del infiltrado puede ser variable tanto en el FCAS como en la urticaria, por lo que la biopsia puede ser considerada de gran ayuda en el diagnóstico, pero no es patognomónica.
Clínica asociadaAdemás de la clínica cutánea descrita es característica de esta enfermedad la aparición en los brotes de fiebre, escalofríos, inyección conjuntival, sudoración, mareos, artromialgias, fatiga y cefalea. Es muy rara en este síndrome la aparición de amiloidosis secundaria, observándose solo en un 2% de los casos. De forma concomitante a los brotes se ha visto que estos pacientes presentan en sangre periférica una marcada leucocitosis.
Síndrome de Muckle-WellsEste síndrome, conocido también como síndrome urticaria-sordera-amiloidosis, es una entidad similar a la anterior, pero con mayor gravedad clínica en los pacientes afectos. Aunque también es habitual su aparición durante la infancia, se ha visto que los pacientes lo inician en un intervalo de edades más variable. Se han descrito múltiples factores desencadenantes, siendo los más frecuentes el calor y el frío.
Clínica cutáneaEl cuadro clínico-patológico es superponible al de los pacientes con FCAS, siendo típica la aparición de una urticaria con un infiltrado inflamatorio de predominio neutrofílico sin edema en la dermis.
Clínica asociadaSon características la aparición de cefalea, meningitis aséptica, inyección conjuntival, edema de papila, artralgias, artritis y cuadros febriles, que son de mayor duración que en FCAS (hasta 36horas), y con intervalos de tiempo entre episodios más irregulares. Destaca la aparición de una sordera neurosensorial progresiva, que comienza en la infancia y puede evolucionar hacia una sordera completa. La amiloidosis secundaria es frecuente, presentándose entre un 25-33% de los pacientes, con habitual afectación renal2. Durante los brotes las alteraciones características en los análisis son un aumento de VSG y PCR, trombocitosis, anemia y leucocitosis con neutrofilia.
Neonatal-onset multisystem inflammatory disease/Chronic infantile neurological cutaneous articular syndromeNOMID es la más grave de las criopirinopatías. Los primeros síntomas suelen aparecer poco después del nacimiento. Se ha descrito que en un 70% de los pacientes las lesiones comienzan en el periodo neonatal, y casi en el 100% antes de los 6 meses de edad.
Muchos de los casos son esporádicos y la mutación en el gen NLRP3 en línea germinal solo está presente en un 55-60% de los pacientes, lo que sugiere una importante heterogeneidad genética, con una gran variedad de mutaciones somáticas3,4. Con motivo de esta gran heterogeneidad, algunos autores proponen un cribado previo a la petición de estudios genéticos cuando se sospecha una criopirinopatía, proponiendo como posibles criterios el haber presentado al menos 3 episodios recurrentes de fiebre moderada y urticaria, una edad de comienzo de la enfermedad <20 años y la presencia de niveles elevados de PCR4.
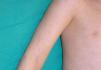
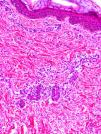
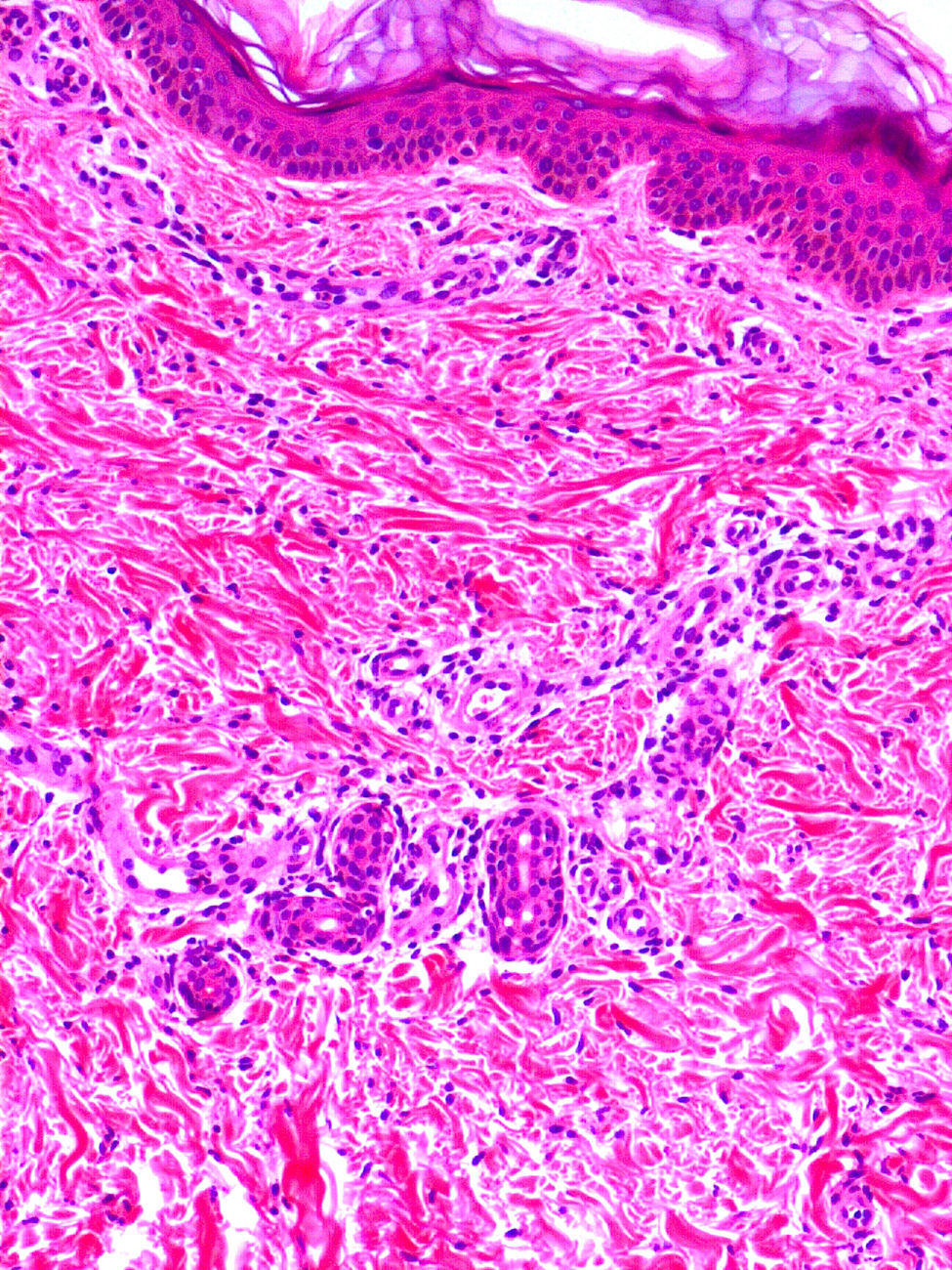
Clínica cutáneaEn estos pacientes también es característica la presencia de una erupción urticariforme migratoria y no pruriginosa, destacando que a diferencia de lo que ocurre en las otras criopirinopatías las lesiones permanecen durante toda la vida y no se relacionan con desencadenantes claros, como el frío (fig. 2). La biopsia de estas lesiones muestra un infiltrado inflamatorio mixto, compuesto de linfocitos, neutrófilos y eosinófilos, con distribución perivascular superficial y profunda, junto a una hidradenitis neutrofílica ecrina5 (fig. 3).
Clínica asociada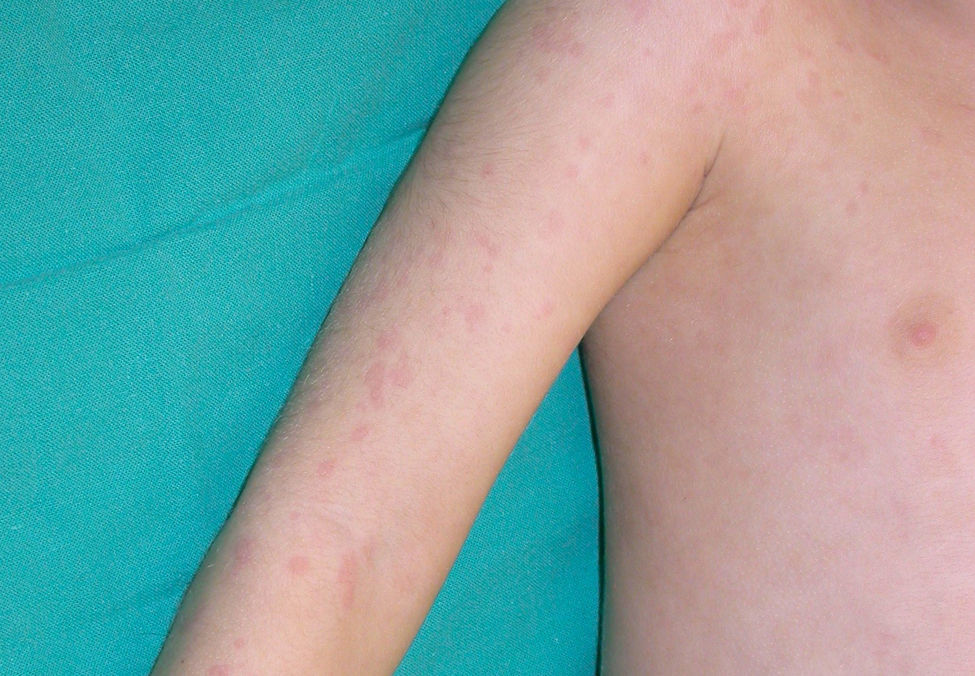
En estos pacientes la clínica asociada es más grave y tiene consecuencias importantes para el desarrollo neurológico. Son frecuentes los episodios cortos de fiebre recurrente, la presencia de linfadenopatías, clínica articular, hepatoesplenomegalia, amiloidosis secundaria, afectación neurológica con atrofia cerebral, retraso mental, meningitis aséptica crónica neutrofílica, convulsiones, hemiplejía transitoria, sordera neurosensorial, cefaleas matutinas, uveítis anterior, aumento de la presión intracraneal, papiledema y ceguera6. Además, presentan un fenotipo facial típico con aplanamiento del puente nasal, macrocefalia, abombamiento frontal e hipertelorismo ocular. La gravedad de la artropatía es variable, siendo muy grave en la mitad de los pacientes antes del primer año de vida y pudiendo simular en los estudios radiológicos tumores óseos secundarios a la osificación en cartílagos de crecimiento y epífisis. En suero destacan la elevación de los reactantes de fase aguda con presencia de leucocitosis, trombocitosis, eosinofilia e hipergammaglobulinemia.
Síndromes hereditarios asociados a fiebres periódicasEn este grupo hemos incluido un conjunto de síndromes autoinflamatorios que se caracterizan por la aparición de una erupción cutánea inespecífica a modo de máculo-pápulas o placas eritematosas que se acompañan con frecuencia de dolor abdominal y cuadros febriles recurrentes, con una duración variable de unos a otros. Esta variabilidad temporal de la fiebre constituye una de sus principales características diferenciales. Los síndromes incluidos son, por un lado, la fiebre mediterránea familiar (FMF), el síndrome de Marshall o síndrome de fiebre recurrente con estomatitis aftosa, faringitis y adenitis y la deficiencia de mevalonato quinasa (o síndrome de hiperinmunoglobulinemia D [HIDS]), en los que la fiebre suele persistir menos de una semana; por otro lado, en el síndrome periódico asociado a factor de necrosis tumoral alfa (TRAPS) la fiebre puede prolongarse incluso varias semanas; también se discutirán en este epígrafe los síndromes PLAID/APLAID, el síndrome AISLE y el síndrome NAIAD.
Fiebre mediterránea familiarLa FMF es un síndrome autoinflamatorio que se transmite de forma autosómica recesiva. Se produce por mutaciones en el gen MEFV, localizado en el cromosoma 16p13, que codifica la proteína pirina/marenostrina. Hasta el momento han sido descritas más de 200 mutaciones para el gen MEFV, siendo las mutaciones en homozigosis en M694V las que asocian una mayor gravedad de la enfermedad. Sin embargo, el diagnóstico es fundamentalmente clínico ya que, en un porcentaje de pacientes con clínica típica de FMF no se encuentra la mutación, debido probablemente a la implicación de otros factores en su desarrollo y a la posible existencia de mutaciones localizadas en otros genes.
Clínica cutáneaLos pacientes presentan de forma característica una placa erisipeloide eritematoedematosa y dolorosa bien delimitada, unilateral o bilateral, localizada en la cara anterior de las extremidades inferiores. Es más frecuente que aparezca por debajo de las rodillas y en el dorso del pie, con un diámetro mayor que no suele sobrepasar los 15cm y con tendencia a recurrir en la misma localización anatómica. Se observan además lesiones purpúricas en la cara, el tronco y las extremidades. Otras manifestaciones cutáneas que aparecen con mayor frecuencia en estos pacientes que en la población general son la púrpura de Schönlein-Henoch (5% de los niños) y la poliarteritis nudosa. Es importante destacar que en la mayoría de los casos de pacientes con FMF la participación cutánea es muy variable y a menudo ausente. La biopsia cutánea de estas lesiones suele mostrar un infiltrado de predominio neutrofílico con cariorrexis.
Clínica asociadaDe forma habitual se manifiesta antes de los 30 años, con la aparición de episodios recurrentes de fiebre elevada (38,5° a 40°C) que se acompañan de astenia grave, monoartritis de grandes articulaciones, principalmente monoartritis con afectación predominante de las extremidades inferiores y dolor abdominal agudo con posible peritonitis. Estos episodios tienen una duración media de uno a 3 días. Además de peritonitis, pueden presentar otras serositis como pleuritis y pericarditis que ocasionan dolor torácico, además de dolor escrotal por inflamación de la túnica vaginal7. La afectación neurológica es inusual, estando descrita la aparición de meningitis en raras ocasiones, durante los episodios agudos, sin cronificación posterior. Las alteraciones serológicas características del episodio agudo incluyen la leucocitosis, con aumento de reactantes de fase aguda, VSG, PCR y fibrinógeno. El aumento de VSG y PCR se mantiene elevado entre las crisis, reflejo de una inflamación subclínica que, sin tratamiento, suele derivar hacia una amiloidosis secundaria, que es la complicación más frecuente e importante del síndrome.
Síndrome periódico asociado al receptor del factor de necrosis tumoralEl TNF es una citocina inflamatoria que desempeña un papel fundamental en la pirexia, la caquexia, la producción de otras citocinas, la expresión de moléculas de adhesión, la activación leucocitaria y la resistencia contra patógenos celulares. El receptor del TNF (TNFR) actúa antagonizando y regulando estas acciones del TNF circulante. TRAPS está producido por una mutación en el gen TNFR que se traduce en la disregulación e hiperfunción del TNF. Este síndrome se hereda con un patrón autosómico dominante, con presencia de mutaciones en el gen TNFRSF1A, que codifica para el receptor i del TNF (también conocido como p55 o CD120a). La gran mayoría de mutaciones identificadas, que son cambios puntuales de un nucleótido por otro en el gen TNFRSF1A, se localizan en los exones 2, 3 y 4 del gen, que codifican para los dominios extracelulares del receptor i del TNF. También se han descrito mutaciones de novo en algunos pacientes.
Clínica cutáneaSe ha descrito la presencia de lesiones cutáneas en un 69-87% de los pacientes con este síndrome. La manifestación más frecuente (40%) se presenta como una placa eritematosa de crecimiento centrífugo y características migratorias, que asienta sobre zonas de mialgia, motivo por el cual también ha recibido el nombre de «eritema doloroso». El tiempo de migración de la placa, de proximal a distal, puede variar desde minutos a varios días y a menudo se desplaza de forma concomitante con la mialgia. En otras ocasiones las lesiones aparecen en forma de placas urticariformes o de una erupción eritematosa máculo-papulosa generalizada que puede evolucionar a placas anulares o serpiginosas, dejando con frecuencia tras su resolución una marcada púrpura equimótica. El edema periorbitario y la conjuntivitis son características que pueden indicar el diagnóstico de TRAPS cuando los hallazgos clínicos asociados son consistentes. La biopsia cutánea muestra un infiltrado de células mononucleares de disposición perivascular e intersticial. En raras ocasiones se ha descrito la presencia de vasculitis leucocitoclástica y paniculitis recurrente.
Clínica asociadaLas manifestaciones clínicas se inician con mayor frecuencia durante la infancia y la adolescencia (con una edad media de 10 años al diagnóstico), pero pueden aparecer desde el primer año de vida hasta la sexta década. Es característica la aparición de episodios febriles que pueden prolongarse durante varias semanas, siendo la duración media de unos 14 días. De forma simultánea a los cuadros febriles, es habitual que los pacientes refieran dolor abdominal repentino e intenso que, como sucedía en la FMF, puede confundirse antes del diagnóstico con un abdomen agudo, y alrededor de un tercio de los pacientes son sometidos a cirugía abdominal. Como hemos mencionado, cuadros musculoesqueléticos con mialgia importante se asocian a las lesiones cutáneas, viéndose en el estudio histopatológico de la biopsia profunda una fascitis monocítica que también se puede apreciar en la resonancia magnética. La mitad de los pacientes tienen clínica ocular como conjuntivitis recurrente o uveítis anterior. Otras manifestaciones como artralgias o artritis, pleuritis, pericarditis, dolor escrotal, cefalea, meningitis aséptica, neuritis óptica y alteraciones del comportamiento también se han observado en el TRAPS. La amiloidosis es la complicación más temida que puede ocurrir en hasta el 24% de los pacientes con mutaciones en los residuos de cisteína, y en el 2% de los pacientes con mutaciones no cisteína que no hayan recibido un tratamiento adecuado7. En el análisis sanguíneo durante los episodios febriles se detecta gammapatía policlonal, leucocitosis, trombocitosis y un aumento de VSG, PCR, ferritina, proteína amiloide sérica y del fibrinógeno, que según la gravedad y cronicidad podrían no llegarse a normalizar en los periodos intercrisis.
Deficiencia de mevalonato quinasa-síndrome de hiperinmunoglobulinemia DEl HIDS se puede dividir en 2 entidades: el clásico, asociado con la mutación genética en el gen MVK y su variante, sin esta mutación genética ni evidencia bioquímica de una reducción de la actividad enzimática asociada a la misma. Estas variantes se han relacionado con mutaciones de baja penetrancia en el gen TNFRSF1A, presentando con frecuencia estos pacientes una clínica más larvada. El gen mutado en los HIDS clásicos es el de la mevalonato quinasa, localizado en el cromosoma 12q24. Aunque típicamente los niveles de IgD suelen estar elevados no siempre es así, y cuando lo están, estos no se correlacionan con la gravedad y la frecuencia de los episodios. Por tanto, dado que está descrito que los niveles de IgD pueden encontrarse dentro de la normalidad, parece que no son responsables directos de las manifestaciones del HIDS, sino que aumentan por un proceso autoinflamatorio inicial desencadenado por traumatismos, vacunas, estrés u otros.
Clínica cutáneaHasta en el 80% de los pacientes aparecen lesiones cutáneas, que pueden ser muy heterogéneas. La más frecuente es una erupción maculosa o máculo-papulosa eritematosa, más o menos confluente, de predominio acral. Otras lesiones pueden aparecer en forma de urticaria, pápulo-nódulos eritematosos o petequias y en menor frecuencia como lesiones similares al síndrome de Sweet, de tipo celulitis, púrpura de Henoch-Schönlein, eritema elevatum diutinum y otras vasculitis. De forma no constante algunos presentan úlceras orales y genitales.
Clínica asociadaLa clínica suele iniciarse en los primeros 4 años de vida (hasta en un 80% de los pacientes aparece antes de los 12 meses) con episodios de fiebre y escalofríos que duran de 3 a 7 días. Por lo general, se repiten cada 4 a 6 semanas, a menudo desencadenados por vacunas, traumatismos, cirugía o estrés. También asocian con frecuencia dolor abdominal con diarrea o vómitos, serositis, cefalea, hepatoesplenomegalia, poliartralgias y artritis no erosiva de grandes articulaciones. Es típico que los pacientes con HIDS presenten linfadenopatías cervicales bilaterales de consistencia blanda. En el análisis sanguíneo también hay un aumento de los reactantes de fase aguda, leucocitosis y neutrofilia. Los niveles séricos de IgD están elevados (≥100U/ml) en más de 90% de los pacientes de forma persistente, y el 80% de ellos presenta de forma concomitante aumento de IgA (≥260mg/dl). Sin embargo, además de que los aumentos de IgD no son específicos y se pueden encontrar en otras condiciones autoinflamatorias como FMF y TRAPS, ya se ha mencionado que podrían no encontrarse en el HIDS, en especial cuando son niños menores de 3 años7. Puede haber un aumento de ácido mevalónico en orina, moderado y solo durante el cuadro agudo.
Síndromes deficiencia asociada a anticuerpos PLCG2 y desregulación inmune/ autoinflamación, deficiencia de anticuerpos y disregulación inmune asociado a PLCG2El síndrome PLAID (deficiencia de anticuerpos asociados a PLCG2 y disregulación del sistema inmune), también conocido como urticaria atípica familiar por frío, es un cuadro de reciente descripción asociado a inmunodeficiencia, hipogammaglobulinemia y fenómenos autoinmunitarios, causado por deleciones en el gen PLCG2 (fosfolipasaC, gamma 2). Los pacientes consultan por cuadros de urticaria, eritema, ardor y granulomas tras la exposición al frío, que pueden asociarse con fiebre, infecciones sinusales y pulmonares recurrentes, asma y enfermedades autoinmunes. Entre las alteraciones en los análisis de sangre destacan el aumento de IgE, los niveles bajos de IgM, IgG, IgA y linfocitos B CD19 circulantes junto a la detección de ANA positivos.
El síndrome APLAID (deficiencia de anticuerpos asociados a PLCG2 y disregulación del sistema inmune con fenómenos autoinflamatorios) es un cuadro similar relacionado con trastornos autoinflamatorios causado por mutaciones puntuales en PLCG2, que asocia bronquiolitis, infecciones pulmonares recurrentes, celulitis, artralgias, enterocolitis, leve inmunodeficiencia sin autoanticuerpos y afectación ocular importante con erosiones, ampollas o ulceraciones corneales, hipertensión ocular y cataratas. Las alteraciones en los análisis son similares a las vistas en el PLAID, pero aquí los ANA son negativos. En el síndrome PLAID la mutación resulta en un aumento de la función y señalización de PLCC2, y en el síndrome APLAID la mutación ocasiona un umbral disminuido para la activación de PLCC2. Ambos síndromes tienen expresión cutánea muy variada, desde cuadros urticariformes inducidos por frío a erupciones vesiculopustulosas que empeoran con el calor, y que pueden ser comunes en ambas entidades. Los granulomas fijos son más frecuentes en el PLAID y las erupciones vesiculoampollosas y las placas recurrentes de celulitis estéril son más frecuentes en el APLAID. En estos cuadros el estudio histopatológico muestra un infiltrado inflamatorio denso de distribución intersticial y perivascular en la dermis, compuesto de neutrófilos, linfocitos, histiocitos y eosinófilos, con fenómenos de vasculitis leucocitoclástica con cariorrexis manifiesta. La mutación descrita en el síndrome APLAID ocasiona un aumento del calcio intracelular, que podría actuar como activador del inflamasoma NLRP3 generando IL-1 (lo que explica una respuesta parcial al tratamiento con anti-IL-1). Según las variantes mutacionales se pueden generar cuadros fenotípicos diferentes, tanto cutáneos como sistémicos, unos inducidos por el frío y otros sin estar relacionados con dicha exposición8,9.
Síndrome autoinflamatorio con linfedemaEl síndrome AISLE o síndrome autoinflamatorio con linfedema es debido a mutaciones en el gel MDFIC, que contiene el dominio inhibidor de la familia MyoD.
Clínica cutáneaSu principal manifestación clínica consiste en una erupción urticariforme extensa.
Clínica asociadaCursa con fiebre, acompañada de edema progresivo de escroto y extremidades inferiores. La anatomía patológica muestra un descenso en el número y tamaño de los vasos linfáticos de la zona.
Síndrome de artritis autoinflamatoria y disqueratosis asociadas a NLRP1A este síndrome, descrito por Grandemange, se le conoce por las siglas en inglés de artritis autoinflamatoria y disqueratosis asociadas a NLRP1. Una activación constitutiva de NLRP1 conlleva un aumento de la función de la caspasa 1, y con ello un incremento de la producción de IL18.
Clínica cutáneaPresentan pápulas eritematomarronáceas hiperqueratósicas de aspecto espinuloso en el tronco y las extremidades. La disqueratosis es el hallazgo fundamental y sus manifestaciones cutáneas son similares a las lesiones observadas en el frinoderma, enfermedad causada por déficit de vitamina A.
Clínica asociadaLos pacientes suelen presentar como clínica acompañante episodios febriles recurrentes y artritis.
Síndromes pustulososDeficiencia del antagonista del receptor de la interleucina-1Se producen mutaciones en el IL1RN (antagonista del receptor de la IL-1), dando lugar a una proteína truncada de menor tamaño que carece de actividad antagonista de IL-1, lo que activa la cascada inflamatoria mediada por IL-1 con importante afectación cutánea y ósea.
Clínica cutáneaEs típico que desde el nacimiento o en los primeros meses de vida los pacientes presenten placas eritematosas generalizadas con pústulas en la superficie, simulando una psoriasis pustulosa. Puede acompañarse de una descamación difusa ictiosiforme, y con frecuencia se respetan las palmas y las plantas. Son comunes las aftas o ulceraciones en la mucosa oral y los cambios ungueales en forma de pits o anoniquia. Las biopsias cutáneas muestran paraqueratosis con pústulas subcórneas o espongiformes de neutrófilos en la epidermis, e infiltración dérmica neutrofílica con afectación variable del folículo piloso, los vasos sanguíneos y las glándulas ecrinas, con una marcada sobreexpresión de IL-17 en técnicas inmunohistoquímicas10.
Clínica asociadaLos brotes suelen acompañarse de fiebre, conjuntivitis, infiltrados pulmonares con distrés respiratorio, episodios trombóticos y alteraciones óseas características como periostitis, osificaciones heterotópicas, fusión vertebral cervical y episodios crónicos de osteomielitis multifocal estéril. Esta última, que con frecuencia afecta las epífisis de los huesos largos, puede ocasionar retraso del crecimiento. En el análisis sanguíneo hay un aumento de reactantes de fase aguda, anemia crónica leve y leucocitosis con neutrofilia sin fiebre asociada. Si la respuesta autoinflamatoria es grave puede ocasionar fracaso multiorgánico y exitus10. Además de su relación con partos pretérmino se han descrito mutaciones que ocasionan afectación multiorgánica intraútero y muerte11.
Deficiencia del antagonista del receptor de la interleuquina-36Las mutaciones identificadas que causan déficit en el antagonista del receptor de IL-36 (IL-36 RN) se asocian con casos tanto familiares (herencia autosómica recesiva) como esporádicos de psoriasis pustulosa generalizada (PPG), y se ha denominado síndrome de la deficiencia del antagonista del receptor de la interleuquina-36 (DITRA).
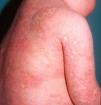
Clínica cutáneaLas lesiones cutáneas en el DITRA se inician de forma aguda como una erupción pustulosa generalizada sobre una base eritematosa12 (fig. 4), muy similar a un brote de psoriasis pustulosa generalizada (PPG), que evoluciona en brotes, con posterior descamación difusa superficial. En algunos casos se puede presentar como una psoriasis vulgar o como lesiones pustulosas de localización acral con destrucción ungueal en forma de acrodermatitis continua. Muchos de los pacientes que presentan brotes de psoriasis pustulosa generalizada, sin clínica previa de psoriasis vulgar, podrían presentar esta mutación genética del DITRA. La frecuencia de los brotes es muy variable entre pacientes, cronificándose en algunos de ellos como placas eritematosas sin pústulas12. La histopatología muestra las características típicas de la PPG con paraqueratosis, acantosis psoriasiforme y pústulas espongiformes, presentando en el infiltrado un predominio de linfocitos T CD8, CD3 y macrófagos.
Clínica asociada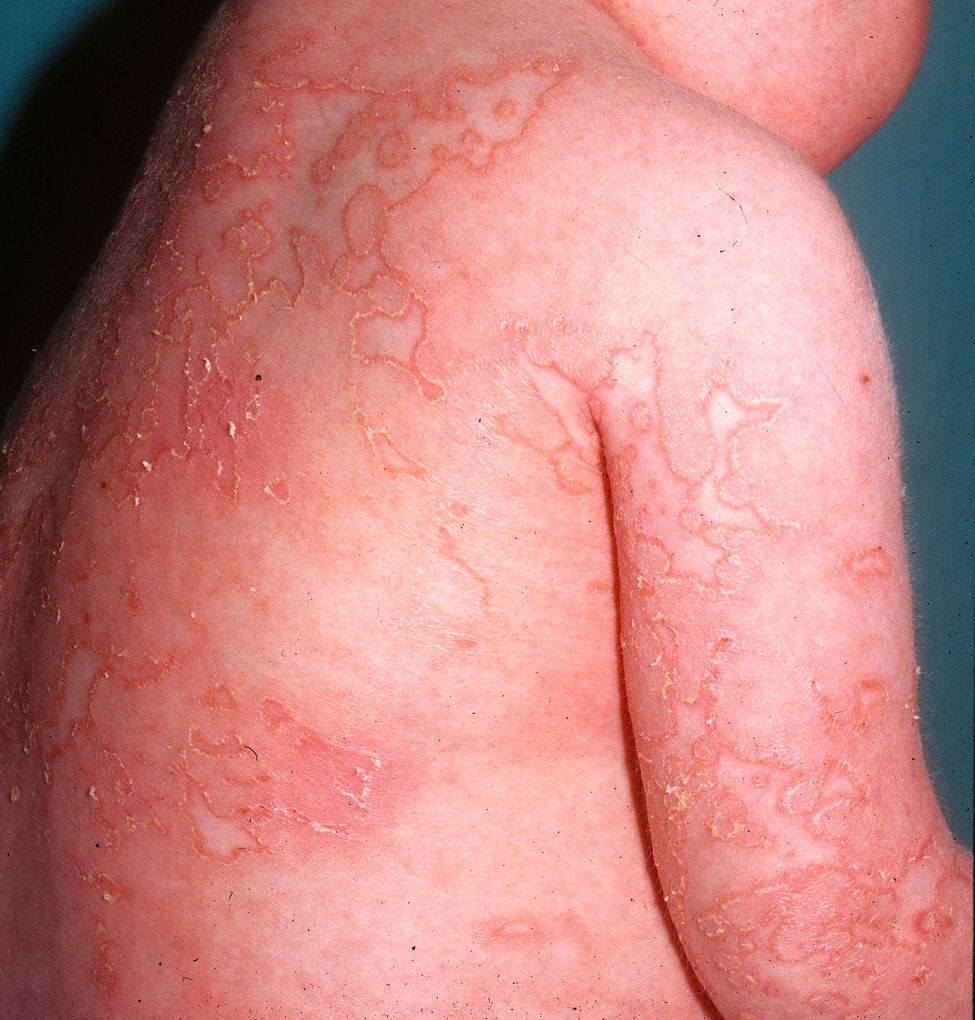
Los brotes cutáneos tienen una frecuencia variable y se asocian con fiebre elevada, malestar y astenia, sin afectación de otros órganos a nivel sistémico. Por lo general estos brotes comienzan en la infancia, pero también existen algunos casos de inicio en el adulto. Se han identificado múltiples desencadenantes como infecciones bacterianas y virales, menstruación, embarazo o fármacos, entre otros. En el DITRA es más llamativa la fiebre elevada y el mal estado general asociado, pero la afectación sistémica es menos importante que en la deficiencia del antagonista del receptor de la interleucina-1, implicando en la mayoría de los casos solo la piel. En el análisis destaca la elevación de los reactantes de fase aguda y el lactato, presentando niveles bajos de albúmina, cinc y calcio.
Síndrome pyogenic sterile arthritis, pyoderma gangrenosum, acneEl síndrome pyogenic sterile arthritis, pyoderma gangrenosum, acne (PAPA), también llamado artritis recurrente familiar, es una enfermedad autosómica dominante poco frecuente que se caracteriza por la triada de artritis piógena, pioderma gangrenoso y acné quístico. La mutación genética está localizada en el cromosoma 15 q24-25.1, que codifica la proteína de interacción prolina/serina/treonina fosfatasa 1 (PSTPIP1), con una penetrancia incompleta y expresión muy variable13. Se han presentado síndromes PAPA con idénticas manifestaciones clínicas, en los que no se ha encontrado la mutación genética, ya que se están descubriendo nuevas mutaciones no descritas con anterioridad14,15. La PSTPIP1 es una proteína del citoesqueleto expresada sobre todo por las células hematopoyéticas, que modula la activación de las células T, la organización del citoesqueleto y la liberación de interleuquina-1β (IL-1β). La mutación genera una sobreproducción de IL-1β.
Clínica cutáneaEs característico que se inicie en la infancia y que empeore de forma importante al llegar a la pubertad. Consiste en formas graves de acné quístico, con fenómeno de patergia y úlceras recurrentes estériles con bordes violáceos sobreelevados periféricos, muy similares a las observadas en el pioderma gangrenoso. También se ha reportado su asociación con psoriasis y rosácea15.
Clínica asociadaEn ocasiones se ha descrito la aparición de fiebre, que aparece de manera no constante y sin ningún tipo de patrón y que se asocia con los episodios recurrentes de artritis erosiva estéril. Esta artritis puede ser espontánea o desencadenada tras un traumatismo menor, y aunque suele disminuir en frecuencia después de la pubertad, en ocasiones persiste en la edad adulta provocando una importante discapacidad. Otras manifestaciones clínicas menos frecuentes son la otitis recurrente, la papilomatosis faríngea, la linfadenopatía, la esplenomegalia, la trombocitopenia, la hipergammaglobulinemia, la anemia hemolítica y la pancitopenia inducida por sulfonamida.
Síndrome de MajeedEste síndrome, al igual que la deficiencia del antagonista del receptor de la interleucina-1, se caracteriza porque hay una afectación ósea importante y presenta una buena respuesta a los antagonistas de la IL-1, hecho que apoya la hipótesis patogénica dependiente de la IL-1β y pone de manifiesto la importante relevancia de esta citoquina en la inflamación ósea16. Sus principales manifestaciones son una osteomielitis multifocal crónica recurrente, una dermatosis neutrofílica y una anemia diseritropoyética congénita con microcitosis. Se han encontrado en los pacientes con este síndrome diferentes mutaciones homocigóticas en el gen LPIN2, que codifica una proteína que modula la transcripción de coactivadores que regulan genes del metabolismo lipídico.
Clínica cutáneaNo es típica ni característica la afectación cutánea en el síndrome de Majeed, siendo más frecuentes los cuadros pustulosos. También se han descrito dermatosis similares al síndrome de Sweet y a la psoriasis.
Clínica asociadaCada exacerbación de la osteomielitis se acompaña de fiebre alta, dolor y tumefacción de las grandes articulaciones. Los focos de osteomielitis son más frecuentes en las clavículas, el esternón y los huesos largos, siendo más rara la afectación de los cuerpos vertebrales y la mandíbula. Esta inflamación crónica ocasiona retraso en el crecimiento, con baja estatura y contracturas en flexión, pudiendo objetivarse en las radiografías lesiones osteolíticas y zonas de esclerosis.
Síndrome autoinflamatorio asociado a pirinas con dermatosis neutrofílicaEste nuevo síndrome autoinflamatorio, que implica una dermatosis neutrofílica pustulosa familiar, fue descrito por Masters en el octavo congreso internacional de FMF y enfermedades autoinflamatorias sistémicas en 2015. Se hereda de forma autosómica dominante y es debido a mutaciones monoalélicas en el gen MEFV diferentes a las implicadas en la fiebre mediterránea familiar, ya que estas mutaciones afectan a una región muy conservada de la proteína pirina, que constituye el punto de unión entre esta y una proteína inhibitoria conocida como 14-3-3.
Clínica cutáneaEs característica la presencia de múltiples pústulas faciales y lesiones pioderma gangrenoso-like17.
Clínica asociadaEl cuadro cutáneo suele acompañarse de fiebre, artromialgias con miositis y elevación de los reactantes de fase aguda.
Síndromes con ulceraciones cutáneo-mucosasSíndrome de fiebre periódica con estomatitis aftosa, faringitis y adenitisEl síndrome de fiebre periódica con estomatitis aftosa, faringitis y adenitis, también conocido como síndrome de Marshall, es el más común de todos los síndromes de fiebres periódicas. Se caracteriza por ser esporádico en la mayoría de los casos y por su resolución espontánea antes del final de la primera década de la vida. Los episodios tienen una periodicidad aproximada de uno al mes, sin predominio estacional, y se autolimitan en unos 3 a 6 días. No se conocen todavía las alteraciones genéticas asociadas a este síndrome, aunque se han descrito casos con cierta agregación familiar.
Clínica cutáneaLo más habitual es que los pacientes presenten pequeñas úlceras aftosas en escaso número en los labios o en la mucosa oral que curan sin dejar cicatriz, acompañadas de un eritema inespecífico urticariforme generalizado de intensidad variable en un pequeño porcentaje de pacientes.
Clínica asociadaEs frecuente que aparezcan faringoamigdalitis agudas de repetición con cultivos negativos y adenopatías cervicales. Pueden acompañarse además de síntomas constitucionales, dolor abdominal, cefalea, artralgias, tos, náuseas o diarrea. Al igual que los demás síndromes de fiebres periódicas presentan elevación de los reactantes de fase aguda en el brote agudo.
Síndrome de fiebre periódica, inmunodeficiencia y trombocitopeniaEl síndrome PFIT o fiebre periódica, inmunodeficiencia y trombocitopenia es un nuevo síndrome autoinflamatorio descrito por Brogan et al.
Clínica cutáneaLa manifestación cutánea más relevante es la aparición de graves úlceras orales que conducen a cicatrices deformantes y microstomía.
Clínica asociadaLos pacientes suelen presentar, además de las úlceras orales, fallo de medro, infecciones de repetición y trombocitopenia.
Síndrome autoinflamatorio Behçet-like asociado con haploinsuficiencia A20Zhou et al. han publicado la presencia en 6 familias no relacionadas, con una mutación no descrita, que implica la pérdida de función en el gen TNFAIP3. Esta mutación conduce a una haploinsuficiencia que provoca un cuadro de inflamación sistémica de inicio precoz. La mutación provoca una degradación aumentada del IkBα que conlleva una translocación de NFkB p65, con una expresión aumentada de las citoquinas proinflamatorias estimuladas por este factor de transcripción18.
Clínica cutáneaLa clínica es muy similar a la observada en la enfermedad de Behçet de inicio pediátrico con ulceraciones orales y genitales.
Clínica asociadaLos pacientes suelen presentar como clínica acompañante fiebre, mal estado general e inflamación ocular, junto a artralgias y artritis de inicio temprano.
Síndrome de BehçetLa enfermedad de Behçet se clasifica como una vasculitis sistémica que puede afectar a cualquier vaso. Es endémica de países del Este del Mediterráneo (ruta de la seda), países del Este y centro de Asia y está asociada con el HLA-B51.
Clínica cutáneaLa clínica cutánea es heterogénea con lesiones de eritema nudoso, pústulas, úlceras necrosantes, tromboflebitis superficial y vasculitis, lesiones similares al pioderma gangrenoso o síndrome de Sweet, entre otras, presentando un fenómeno de patergia positivo. Las úlceras aftosas orales recidivantes son la primera manifestación en un elevado porcentaje de casos; suelen ser múltiples, dolorosas y curar sin dejar cicatriz en contraposición con las úlceras genitales, que sí dejan unas características cicatrices planas. Es característico encontrar en el estudio histopatológico fenómenos de vasculitis neutrofílica y/o trombosis.
Clínica asociadaLa afectación ocular es la principal causa de morbilidad. La más relevante es la uveítis posterior, pero también presentan uveítis anterior e iridociclitis con hipopion. En ocasiones se acompaña de artritis y de un gran abanico de manifestaciones secundarias a la vasculitis sistémica, como trombosis arteriales y venosas, aneurismas, clínica digestiva y neurológica.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses


