



El síndrome de Hay-Wells, también conocido como síndrome AEC (anquilobléfaron, displasia ectodérmica y hendidura palatina, herencia mendeliana en el hombre [OMIM] 106260) es un trastorno genético dominante autosómico raro asociado a una mutación heterocigota en el gen TP63. El síndrome AEC se define por la presencia de anomalías ectodérmicas en piel, dientes, cabello y uñas, siendo también característica la fusión de párpados y hendiduras faciales1,2. Otros trastornos autosómicos dominantes del desarrollo vinculados con el gen TP63 incluyen el síndrome de Rapp-Hodgkin (RHS; OMIM 129400), el síndrome ectrodactilia-displasia ectodérmica-hendiduras orofaciales (síndrome EEC; OMIM 604292), el síndrome acro-dermato-ungueal-lacrimal-dental (síndrome ADULT; OMIM 103285), el síndrome de miembros y mamas (LMS; OMIM 603543) y el síndrome de malformación de la mano hendida/pie hendido (síndrome SHFM; OMIM 605289). Como las características clínicas e incluso moleculares suelen solaparse se ha sugerido que algunos de estos síndromes son representativos de un espectro variable del mismo trastorno genético3.
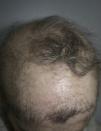
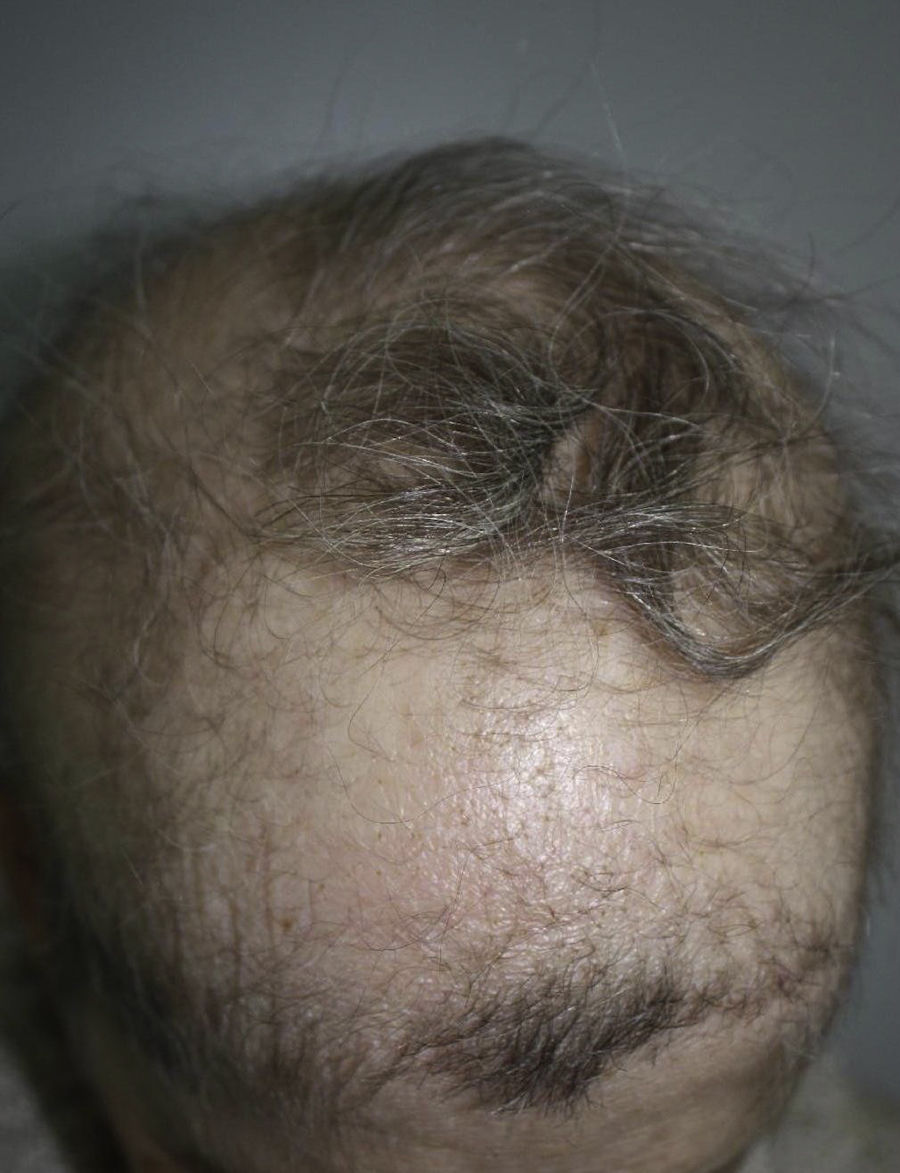
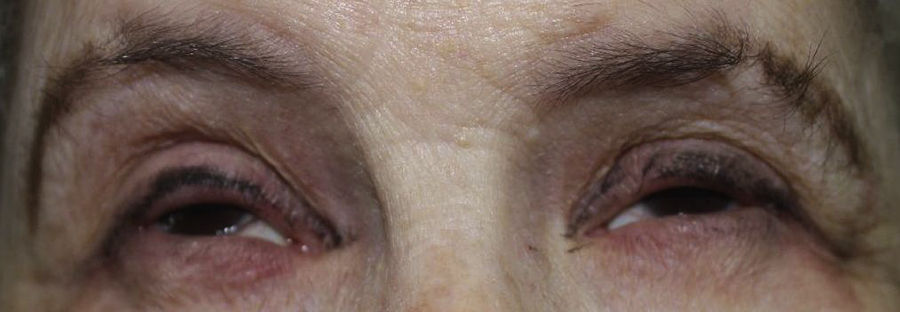
Una mujer de 57 años con antecedentes personales de numerosas intervenciones quirúrgicas oftalmológicas fue derivada a su oftalmólogo para someterse a una biopsia de la mucosa bucal a fin de descartar penfigoide cicatricial. La exploración física reveló alopecia en parches de cuero cabelludo, cejas y párpados (fig. 1), distrofia ungueal, hipodontia e hipohidrosis, todas estas afecciones presentes desde la infancia o el nacimiento. También se observó un descenso del diámetro ocular y una cierta tendencia a la adhesión de los bordes ciliares de los párpados. Esta situación provocó una fotosensibilidad grave (fig. 2). Más recientemente, la paciente ha presentado hiperqueratosis palmoplantar. No se observó presencia de labio ni paladar hendidos.
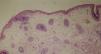
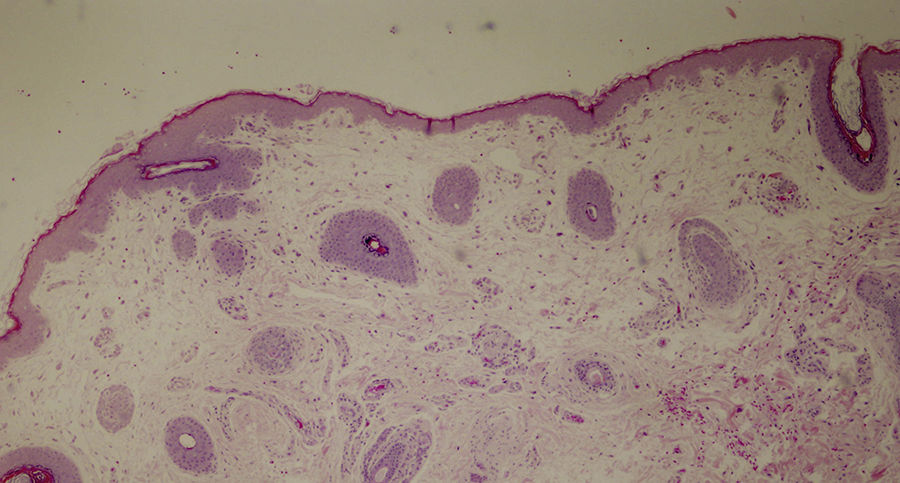
Se propuso un diagnóstico de síndrome de displasia ectodérmica, más concretamente de síndrome de Hay-Wells o de AEC. Una biopsia del cuero cabelludo reveló la presencia de estructuras pilosas rudimentarias, algunas de las cuales dieron lugar a cabello de tipo velloso y ausencia total de glándulas sebáceas (fig. 3).
El estudio genético mostró la presencia de una mutación heterocigota Arg243Trp del gen TP63 (c.727C>T), anteriormente descrita en la Base de Datos de Mutaciones Genéticas Humanas, solo asociada a un genotipo del síndrome EEC pero nunca a un genotipo del síndrome AEC. Se ofreció asesoramiento genético a miembros de su familia, en varias ocasiones, pero la paciente declinó la oferta.
El gen TP63 es un miembro de la familia genética TP53 que codifica la expresión de p63, una molécula clave en el desarrollo craneofacial y de las extremidades en la diferenciación de la piel y la carcinogénesis. Su estructura consiste en 5 dominios, incluido el de transactivación, el de unión al ADN, el de oligomerización, el dominio con motivo alfa estéril (SAM) y el de inhibidores de transactivación. Las mutaciones de Tp63 que asocian ectrodactilidad suelen localizarse en el dominio de unión al ADN, al igual que ocurre con el síndrome EEC, mientras que el síndrome AEC y otras mutaciones sin ectrodactilidad suelen venir provocadas por mutaciones del dominio p63 SAM4–6. Las mutaciones del dominio de unión al ADN asociadas a un fenotipo del síndrome AEC son muy raras, como en el caso aquí descrito, por eso estas mutaciones han de mantenerse bajo la categoría de «importancia incierta».
La variabilidad clínica es una de las señas de identidad del síndrome AEC4. Los síntomas más significativos son displasia ectodérmica con cabello áspero, tieso y escaso, uñas distróficas, hipohidrosis de carácter leve, infecciones del cuero cabelludo, anquilobléfaron filiforme adherente, hipodontia, hipoplasia maxilofacial y labio/paladar hendidos. Otras características incluyen hiperqueratosis palmoplantar, nariz grande, trastorno de pigmentación cutánea o deformidades del oído1,7,8.
El principal diagnóstico diferencial debe establecerse con el síndrome RHS y EEC. El síndrome AEC difiere de las otras enfermedades asociadas a la mutación del gen TP63 en la gravedad del fenotipo de piel, la ausencia de ectrodactilidad y, sobre todo, en la presencia de anquilobléfaron. El labio/paladar hendido es otra característica propia del síndrome de AEC que comparte con el síndrome EEC, RHS y LMS, pero no con el síndrome ADULT, y tampoco es típica del SHFM.
Se ha propuesto que los síndromes RHS y AEC representan un espectro variable del mismo trastorno genético3,9, ya que se solapan en las características clínicas y moleculares, y en algunos casos comparten las mismas mutaciones. La única variación fenotípica entre ambos síndromes es el anquilobléfaron, que se presenta en más del 58% de casos de síndrome AEC3.
La presencia de displasia ectodérmica asociada a anquilobléfaron se ha descrito en otros síndromes como el CHANDS (cabellos rizados-anquilobléfaron-displasia ungueal) y el de Rosselli-Giulienetti, en el diagnóstico diferencial del síndrome AEC, si bien su modo de herencia es autosómico revesivo10.
El tratamiento del síndrome AEC es sintomático. El asesoramiento genético es útil tanto para el propio sujeto como para los miembros de su familia afectados. El pronóstico es favorable, con mejora progresiva de las lesiones cutáneas7,8.
En conclusión, describimos un caso del síndrome AEC con una mutación previamente, solo asociada a un fenotipo del síndrome EEC. Esto sugiere que todos los trastornos asociados al gen TP63 podrían ser el resultado de la variabilidad fenotípica dentro de un espectro de una sola enfermedad genética.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


