

Hay-Wells syndrome, also known as AEC syndrome (ankyloplepharon-ectodermal dysplasia-clefting syndrome, Online Mendelian Inheritance in Man [OMIM] 106260) is a rare, autosomal dominant genetic disorder, associated with a heterozygous mutation in the TP63 gene. AEC syndrome is defined by ectodermal abnormalities of the skin, teeth, hair, and nails, in combination with characteristic eyelid fusion and facial clefting.1,2 Other distinct autosomal dominant developmental disorders have been associated with the TP63 gene, including Rapp-Hodgkin syndrome (RHS; OMIM 129400), ectrodactyly-ectodermal dysplasia-cleft lip/palate syndrome (EEC syndrome; OMIM 604292), acro-dermato-ungual-lacrimal-tooth syndrome (ADULT syndrome; OMIM 103285), limb-mammary-syndrome (LMS; OMIM 603543) and split hand/foot malformation syndrome (SHFM syndrome; OMIM 605289). As clinical and even molecular features commonly overlap, it has been proposed that some of these syndromes represent a variable spectrum of the same genetic disorder.3
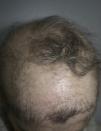
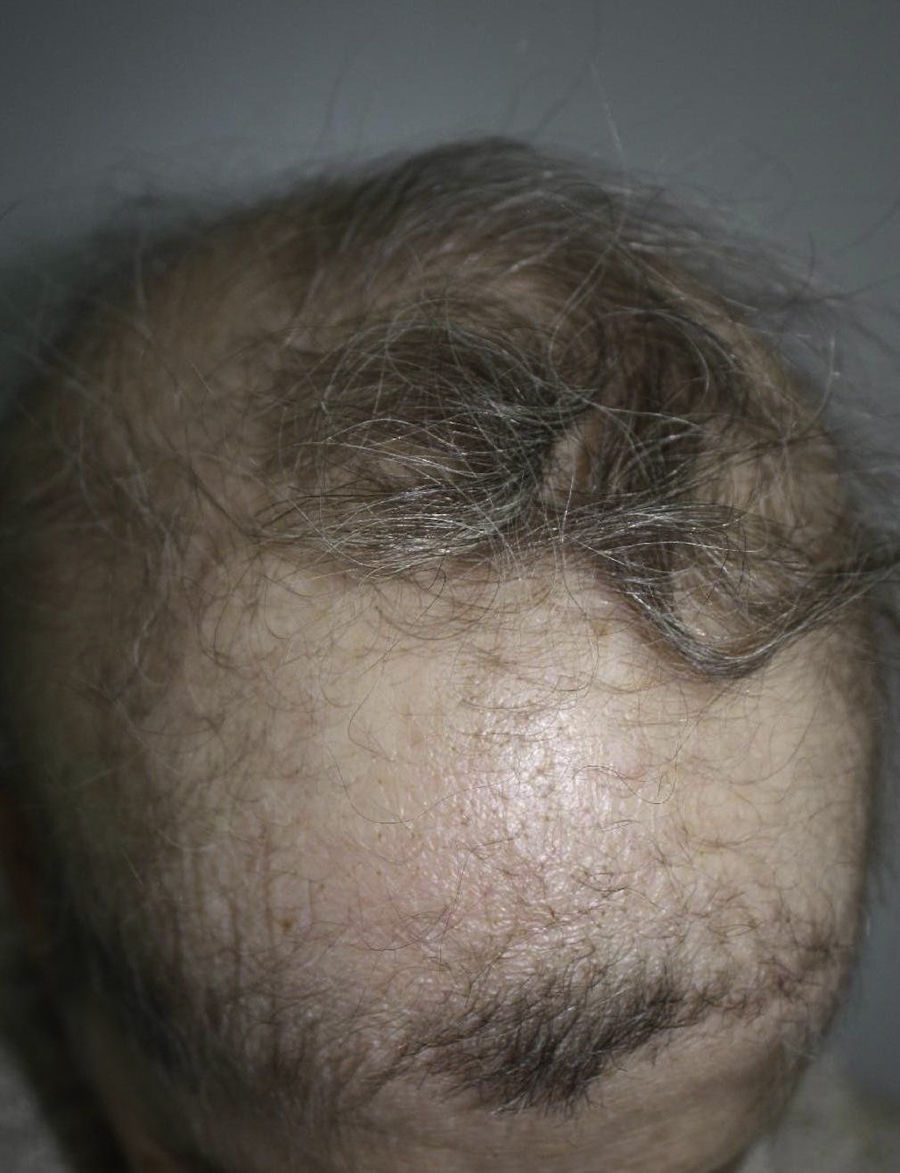
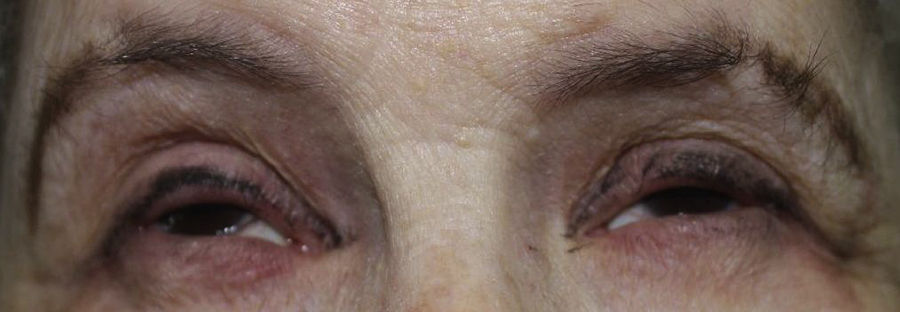
A 57-year-old woman with a personal history of numerous ophthalmologic surgical procedures was referred to her ophthalmologist for a biopsy of buccal mucosa to rule out cicatricial pemphigoid. Physical examination revealed patchy alopecia of the scalp, eyebrows and eyelids (Figure 1), nail dystrophy, hypodontia and hypohidrosis, all these conditions present since childhood or birth. Furthermore, there was a decreased eye diameter and a tendency to adhesion of the ciliary edges of the eyelids. This situation caused severe photosensitivity (Figure 2). More recently, the patient presented palmoplantar hyperkeratosis. Neither cleft lip nor palate was present.
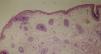
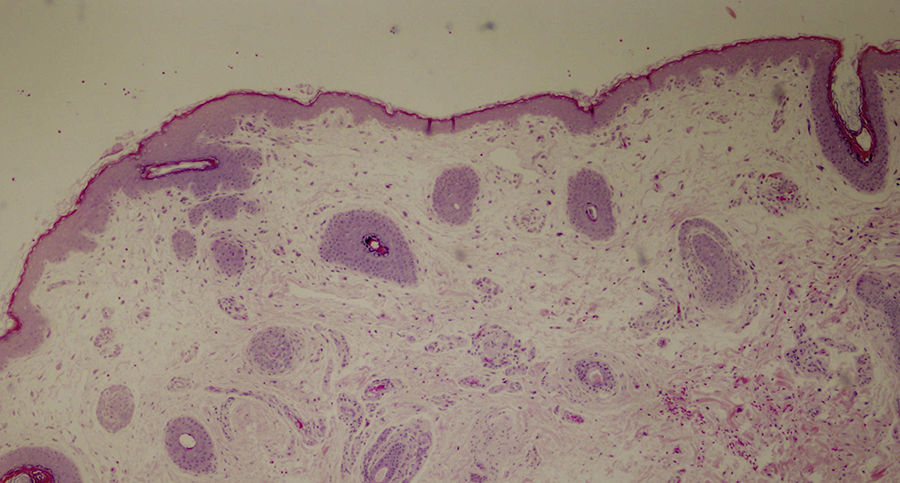
Diagnosis of ectodermal dysplasia syndrome was proposed, more specifically of Hay-Wells (AEC) syndrome. A punch biopsy specimen from the scalp revealed the presence of rudimentary hair structures, some of which gave rise to vellus type hair, and total absence of sebaceous glands (Figure 3).
The patient was offered genetic testing and was found to have an heterozygous Arg243Trp mutation in the TP63 gene (c.727C>T), which was previously reported in the Human Gene Mutation Database only associated with a phenotype of EEC syndrome, but never with a phenotype of AEC syndrome. Genetic counselling in family members was offered on several occasions but the patient always refused.
The TP63 gene is a member of the TP53 gene family that encodes for p63, a key molecule in craniofacial and limb development, skin differentiation and carcinogenesis. Its structure comprises five domains, including transactivation domain, DNA-binding domain, oligomerization domain, sterile-alpha-motif (SAM) domain and the transactivation inhibitors domain. Tp63 mutations associating ectrodactily are usually located in the DNA-binding domain, as occurs in EEC syndrome, whereas AEC syndrome and other mutations without ectrodactily are mostly caused by mutations in the p63 SAM domain.4–6 Mutations in DNA-binding domain associated with a phenotype of AEC syndrome are very seldom reported, as in the case described here, so these mutations must remain under “uncertain significance”.
Clinical variability is one of the hallmarks of AEC syndrome.4 The major symptoms include congenital ectodermal dysplasia with coarse, wiry, sparse hair; dystrophic nails; slight hypohidrosis; scalp infections; ankyloblepharon filiforme adnatum; hypodontia; maxillary hypoplasia; and cleft lip/palate. Other features include palmoplantar hyperkeratosis, broad nose, skin pigmentation disorder or ear deformities.1,7,8
The main differential diagnosis must be established with RHS and EEC syndrome. AEC syndrome differs from the other TP63 mutation-related conditions in the severity of skin phenotype, absence of ectrodactyly and, especially, the occurrence of ankyloblepharon. Cleft lip/palate is also a feature of AEC syndrome that is shared with EEC syndrome, RHS and LMS, but not with ADULT syndrome, and it is not typical of SHFM.
It has been proposed that RHS and AEC syndrome represent a variable spectrum of the same genetic disorder,3,9 as they overlap in clinical and molecular features, as reported in some of the cases of both entities sharing the same mutations. The only phenotypic variation between the two syndromes is ankyloblepharon, which is present in over 58% of reported cases of AEC syndrome.3
The presence of ectodermal dysplasia associated with ankyloblepharon has been reported in other syndromes such as CHANDS (curly hair-ankyloblepharon-nail dysplasia syndrome) and Rosselli-Giulienetti syndrome that should be considered in the differential diagnosis of AEC syndrome, although its mode of inheritance is autosomal recessive.10
Treatment of AEC syndrome focuses on the symptoms present. Genetic counselling is helpful for the individual and family affected. The prognosis of patients with AEC syndrome is favourable, with progressive improvement of cutaneous lesions.7,8
In conclusion, we report a case of AEC syndrome presenting a mutation previously only associated with a phenotype of EEC syndrome, suggesting that all TP63-related disorders may be a result of phenotypic variability within a spectrum of a single genetic condition.
Please cite this article as: Romero-Pérez D, Encabo-Durán B, Ramón-Sapena R. Ampliando el perfil genético del síndrome de Hay-Wells. Actas Dermosifiliogr. 2018;109:560–562.


