










El carcinoma de células de Merkel (CCM) es un tumor poco frecuente, con un curso evolutivo muy agresivo, que con frecuencia origina recidivas locorregionales y metástasis. Asienta predominantemente en zonas fotoexpuestas en pacientes ancianos. Su incidencia se ha cuadriplicado en las últimas décadas debido al envejecimiento de la población y a un mayor diagnóstico gracias al uso de técnicas inmunohistoquímicas. La patogénesis del CCM no está clara, pero la radiación ultravioleta, la inmunosupresión y la presencia del poliomavirus MCPyV en el genoma del tumor parecen desempeñar un papel fundamental en el desarrollo de esta neoplasia.
El objetivo de este artículo es realizar una revisión actualizada sobre los diferentes aspectos de la epidemiología, la etiopatogenia y la clínica del CCM. A su vez, detallamos los aspectos histológicos e inmunohistoquímicos necesarios para su diagnóstico y revisamos la estadificación por su importante trascendencia en el pronóstico de estos pacientes.
Merkel cell carcinoma (MCC) is a rare, highly aggressive tumor, and local or regional disease recurrence is common, as is metastasis. MCC usually develops in sun-exposed skin in patients of advanced age. Its incidence has risen 4-fold in recent decades as the population has aged and immunohistochemical techniques have led to more diagnoses. The pathogenesis of MCC remains unclear but UV radiation, immunosuppression, and the presence of Merkel cell polyomavirus in the tumor genome seem to play key roles. This review seeks to update our understanding of the epidemiology, etiology, pathogenesis, and clinical features of MCC. We also review histologic and immunohistochemical features required for diagnosis. MCC staging is discussed, given its great importance in establishing a prognosis for these patients.
El carcinoma de células de Merkel (CCM), o carcinoma neuroendocrino primario cutáneo, es un tumor cutáneo poco frecuente que afecta a pacientes ancianos y tiene un curso evolutivo muy agresivo, que frecuentemente origina recidivas locorregionales y metástasis. Debido a un incremento en su incidencia y al descubrimiento de un virus en su patogenia, el interés en el CCM ha aumentado en los últimos años.
El objetivo de este artículo es realizar una revisión actualizada sobre los diferentes aspectos de la epidemiología, la etiopatogenia y la clínica del CCM. Se revisan los aspectos histológicos e inmunohistoquímicos necesarios para su diagnóstico y estadificación actual del paciente según la American Joint Committee on Cancer.
HistoriaInicialmente fue descrito por Toker et al. en 19721 con el nombre de «carcinoma trabecular de la piel», sugiriendo un posible origen glandular. Seis años después estos autores2 identifican mediante un estudio ultraestructural la presencia de gránulos electrodensos en el citoplasma de las células tumorales, proponiendo un origen neuroendocrino, similar a las células de Merkel de la epidermis. Actualmente su origen celular sigue siendo un tema a debate, lo que explica la cantidad de términos empleados para nombrarlo desde que fue descrito: apudoma cutáneo, carcinoma primario de células pequeñas de la piel, carcinoma anaplásico de la piel, carcinoma endocrino de la piel, merkeloma, carcinoma neuroendocrino primario cutáneo y CCM3–5. Todos estos términos reflejan su diferenciación neuroendocrina, la morfología de sus células y su semejanza histológica con el carcinoma de células pequeñas de pulmón.
El nombre «carcinoma neuroendocrino cutáneo» es quizás el que refleja mejor las características inmunohistoquímicas y ultraestructurales del tumor, pero el término más empleado en la literatura, y que se acepta universalmente es CCM o «tumor de Merkel».
EpidemiologíaEl CCM es un tumor muy poco frecuente, pero característicamente muy agresivo, duplicando en mortalidad al melanoma6. Representa menos del 1% de los tumores cutáneos malignos7, sin embargo constituye la tercera causa de muerte por cáncer de piel después del melanoma y del carcinoma epidermoide8.
La incidencia actual en España es desconocida, pero en Estados Unidos ha ido en aumento de manera progresiva, triplicándose en las últimas décadas, de 0,15 a 1,44 por 100.000 habitantes entre 1981 y 20119,10. Este incremento es debido a un mayor conocimiento de la enfermedad por parte del dermatólogo y del patólogo, a una mayor facilidad diagnóstica por el uso de las técnicas inmunohistoquímicas y a un aumento de la población de riesgo a causa de la exposición ultravioleta, la edad avanzada y estados de inmunosupresión11.
El CCM afecta casi exclusivamente a la raza blanca, mientras que es excepcional en la raza negra. Es algo más frecuente en varones que en mujeres en algunos trabajos (1,4:1), pero en otros no encuentran predominio por el sexo7,12.
Este tumor ocurre con mayor frecuencia en personas de edad avanzada, con un pico de incidencia entre la séptima y la octava décadas de la vida13. La edad media al diagnóstico es de 75 años, y solo el 5% de los pacientes son menores de 50 años12, y habitualmente asociados a algún tipo de inmunosupresión. No obstante, se han descrito casos en niños en los que el más joven tenía 7 años14.
Etiopatogenia y población de riesgoEl CCM afecta a individuos de raza blanca, de fototipo claro (i-iii), en lugares fotoexpuestos y asociados a otros tumores cutáneos. Estos datos van a favor de un papel de las radiaciones ultravioletas en su etiopatogenia15,16. Además ha sido descrito tras la irradiación, en pacientes tratados con PUVA terapia o expuestos a arsénico17 y la aparición en áreas de eritema ab igne18. Los pacientes con CCM presentan también otros tumores fotoinducidos cutáneos como carcinoma basocelular, carcinoma espinocelular y melanoma. Estos tumores pueden aparecer antes o después del CCM. Los pacientes con melanoma presentan un riesgo 3 veces mayor que la población general de desarrollar un CCM19.
Esta neoplasia aparece con más frecuencia en personas inmunodeprimidas12,20, especialmente en pacientes con trasplante cardiaco21 o renal22, o que están recibiendo tratamiento inmunosupresor con azatioprina, ciclosporina, ciclofosfamida e inhibidores de mTOr23 y en pacientes con infección por el virus de la inmunodeficiencia humana (VIH)24. Por tanto, la inmunosupresión es un factor de riesgo para su desarrollo, y además, estos casos presentan un curso más agresivo y surgen en edades más tempranas. El periodo medio de aparición tras el trasplante es aproximadamente de 10 años, y en estos pacientes la proporción de melanoma en relación con el CCM es de 1:6 frente a la población general, cuya relación es de 1:65.
En los pacientes con el virus de la inmunodeficiencia humana existe un riesgo 13 veces mayor para el desarrollo del CCM en comparación con la población general. En un estudio realizado en 14 pacientes con VIH y CCM se detectó que la mayoría de los pacientes eran varones y más jóvenes (49 años) que los pacientes con CCM no VIH (70 años)25. A diferencia de lo que ocurre en la población general, el tumor se localiza en áreas no expuestas (35% en nalgas), y la media de supervivencia es de menos de 19 meses tras el diagnóstico.
Infrecuentemente se ha descrito la asociación de CCM en pacientes con enfermedades hematológicas26 (leucemia linfática crónica, linfoma no Hodgking, mieloma múltiple), tumores sólidos27 (pulmón, páncreas, próstata, mama o colon) y enfermedades autoinmunes (artritis reumatoide, espondilitis anquilosante, enfermedad de Crohn y enfermedad inflamatoria intestinal)28,29. De todos ellos la leucemia linfoide crónica es la enfermedad que más frecuentemente se asocia a CCM. Se ha descrito de manera esporádica la asociación con displasia ectodérmica congénita y con la enfermedad de Cowden30.
El descubrimiento más importante relacionado con la patogenia del CCM ha sido la caracterización en el año 2008 de un virus, llamado poliomavirus de células de Merkel (MCV o MCPyV) en el genoma de las células del tumor. Feng et al.31 encuentran el genoma de este virus en el 80% de los CCM analizados (8/10), y solamente en el 8% de controles en diferentes órganos y en el 16% de controles en piel sana. Estudios posteriores han confirmado que aproximadamente el 80% de los CCM tienen el poliomavirus. No obstante, la incidencia de casos que presentan el poliomavirus es variable, y algunos investigadores relacionan esta variabilidad con las diferentes áreas geográficas.
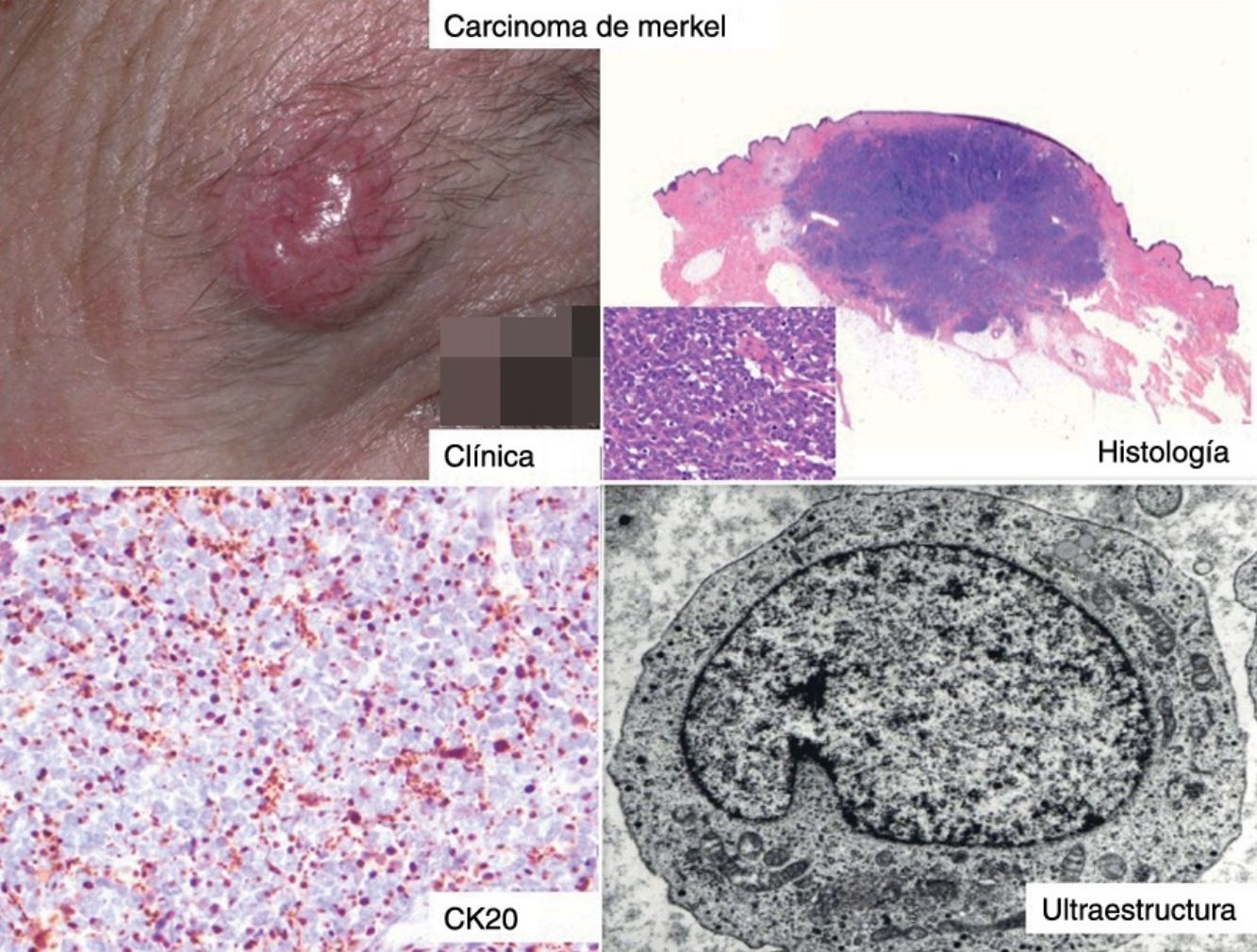
ClínicaEl CCM en la mayoría de los casos se presenta como una tumoración nodular única, firme, de coloración eritematosa, violácea o púrpura, bien delimitada y móvil con respecto a planos profundos. La superficie de la lesión es suave, lisa, brillante, raramente se ulcera y con frecuencia presenta telangiectasias32,33 (fig. 1A). En ocasiones la lesión se presenta como una placa única dérmica o subcutánea o como una placa multinodular (fig. 1B), y de manera menos frecuente como una lesión multifocal o diseminada (fig. 1C)34–36. Se han descrito casos pediculados (fig. 1D)37.

Clínica del carcinoma de células de Merkel. A: lesión nodular eritematosa, bien delimitada, de superficie lisa, brillante con telangiectasias en su superficie. B: placa mal delimitada con múltiples nódulos en superficie. C: carcinoma de células de Merkel diseminado. D: carcinoma de células de Merkel de pequeño tamaño.
La lesión es asintomática y el paciente consulta al médico por el rápido crecimiento del tumor en semanas o meses38. El tamaño medio de la tumoración es de 2-4cm al establecer el diagnóstico5,33,39–41. En lesiones de gran tamaño en ocasiones se observa la presencia de un halo inflamatorio eritematoso peritumoral. En la histología de estas áreas no se observa infiltrado inflamatorio, y sí la presencia de invasión vascular. Por tanto, la presencia de este halo es un signo clínico de mal pronóstico.
El CCM es más frecuente en áreas fotoexpuestas, pero puede asentar en cualquier área cutánea o mucosa. Su localización más frecuente es la cabeza y el cuello en alrededor de un 50%, seguido de las extremidades en un 40% y un 10% en el tronco y en los glúteos32,35,42,43. La región periorbitaria es una localización muy frecuente (10%) (fig. 1A), y en esta localización se suele confundir con un proceso benigno tipo quiste o chalazión. En raras ocasiones se ha descrito en zonas no expuestas como la mucosa genital44,45, perianal y orofaríngea46.
Están publicados casos de CCM localizados en ganglio en ausencia de tumor primario en la piel47. Cabe destacar que estos CCM primarios nodales se encuentran asociados a un mejor pronóstico que los primarios cutáneos con afectación ganglionar48. Además, se han descrito casos de CCM que regresan de manera parcial o total de forma espontánea o tras la realización de una biopsia49,50.
En algunos casos se diagnostican CCM in situ o de pequeño tamaño (Baby Merkel) en el seguimiento de pacientes con carcinomas basocelulares o epidermoides, o como hallazgo incidental en piezas quirúrgicas de otros tumores51–54. En estos casos el pronóstico es mejor.
El CCM puede parecer en su inicio una lesión benigna y demorar así el tratamiento, por lo que se ha definido el acrónimo «AEIOU» (Asintomático, Expansión rápida, Inmunosupresión, mayores de 50 años —Old— y exposición Ultravioleta) en un intento de resumir las características clínicas más importantes de dicho tumor, y de esa forma no retrasar el diagnóstico. La sospecha diagnóstica inicial de CCM generalmente no se plantea como primera opción, solamente en un 1% de todos los casos55.
El diagnóstico diferencial clínico es muy variado e incluye tanto lesiones benignas (quiste epidérmico, dermatofiborma, fibroma, angioma y chalazión) como malignas (carcinoma espinocelular, melanoma amelanótico, metástasis cutáneas de otros tumores, linfomas, carcinoma basocelular, queratoacantoma y tumores anexiales)54.
La dermatoscopia no es característica en el CCM, pero se ha descrito la presencia de estructuras rosadas lechosas en zonas con desorganización arquitectural y la presencia de vasos arboriformes que deben hacer sospechar una lesión maligna, y entre ellas el CCM56,57.
HistologíaMacroscópicamente es un tumor bien delimitado, no encapsulado, firme y homogéneo. Microscópicamente es una lesión dérmica constituida por grupos de células redondas, basófilas, monomorfas, con núcleos grandes vesiculosos con cromatina fina y granular, que se conoce como «en sal y pimienta»58 (fig. 2). Es frecuente observar áreas necróticas, núcleos picnóticos y abundantes figuras mitósicas (fig. 2D). La invasión vascular es común, al igual que la presencia de un infiltrado inflamatorio abundante alrededor del tumor formado por linfocitos y células plasmáticas, y en ocasiones se observa infiltrado inflamatorio intratumoral.
. C: tumor localizado en la dermis constituido por células redondas, basófilas y monomorfas (hematoxilina-eosina, × 100). D: las células son redondas, azules, con escaso citoplasma y núcleo con cromatina fina y granular. En el tumor se observan numerosas figuras mitósicas (hematoxilina-eosina, × 400).")
Estudio histológico. A y B: panorámica de un carcinoma de células de Merkel. Tumor bien delimitado, no encapsulado, nodular (A y B:hematoxilina-eosina, x10). C: tumor localizado en la dermis constituido por células redondas, basófilas y monomorfas (hematoxilina-eosina, × 100). D: las células son redondas, azules, con escaso citoplasma y núcleo con cromatina fina y granular. En el tumor se observan numerosas figuras mitósicas (hematoxilina-eosina, × 400).
Habitualmente existe una zona de separación del tumor con la epidermis (fig. 3), solo se encuentra ulceración de la epidermis en aproximadamente un 15% de los casos y menos de un 10% de los CCM presentan afectación epidérmica (epidermotropismo)42,59. Excepcionalmente se han descrito casos confinados exclusivamente a la epidermis (CCM in situ)53,60 o con mínima afectación dérmica51. En la epidermis puede formar agregados o adoptar un patrón pagetoide que puede imitar a otros tumores de crecimiento intraepidérmico como el melanoma, la micosis fungoide, la enfermedad de Bowen o la enfermedad de Paget61.
; B: la tumoración infiltra ampliamente la dermis (hematoxilina-eosina × 200). C: células atípicas, pequeñas, redondas y basófilas (hematoxilina-eosina × 400). D: infiltración de la tumoración entre los haces de colágeno (hematoxilina-eosina × 200).")
Histología de un carcinoma de células de Merkel con un patrón infiltrativo. A: tumoración en dermis con epidermis conservada (Hematoxilina-eosina × 100); B: la tumoración infiltra ampliamente la dermis (hematoxilina-eosina × 200). C: células atípicas, pequeñas, redondas y basófilas (hematoxilina-eosina × 400). D: infiltración de la tumoración entre los haces de colágeno (hematoxilina-eosina × 200).
En esta tumoración se han descrito diferentes patrones histológicos (patrón trabecular, nodular y difuso) sin implicaciones pronósticas62. El patrón trabecular descrito por Toker en 19721, es poco frecuente y suele estar limitado a la periferia de la lesión. Se caracteriza porque las células se organizan en trabéculas separadas por un estroma fibroso, y estas células se encuentran apiladas dando un aspecto compacto, con formación de estructuras que recuerdan a glándulas y rosetas. El patrón más frecuente es el nodular, y se caracteriza porque las células se disponen formando nódulos sólidos. El aspecto general de este patrón recuerda bastante al de un linfoma. El patrón difuso se caracteriza por un infiltrado de células pequeñas, en sábana, separadas por un estroma abundante. La actividad mitótica es muy alta y existen grandes áreas de necrosis. En este caso el aspecto es similar al carcinoma de células pequeñas del pulmón. Es muy frecuente encontrar varios de estos patrones dentro de un mismo tumor.
De manera excepcional se ha descrito CCM con patrones de diferenciación divergente en el 5% de los casos63. El patrón con diferenciación glandular ecrina64,65 se confirma por la expresión inmunohistoquímica de CK7 y CEA. También se han descrito casos con diferenciación escamosa66, melanocítica63, muscular y otros con patro¿n de «linfoepitelioma»67. Estos casos con infiltracio¿n linfoide se han relacionado con la regresio¿n tumoral. La diferenciación sarcomatosa puede ser de tipo «fibrosarcomatosa»68, «leiomiosarcomatosa»69, «rabdomiosarcomatosa» o «fibroxantoma atípico» con la presencia de células gigantes multinucleadas70. En algunos casos se ha descrito, incluso, CCM con diferenciacio¿n glandular, escamosa y melanoci¿tica en un mismo tumor63. Su capacidad para diferenciarse hacia otras líneas celulares iría a favor de un origen a partir de células madre pluripotenciales71,72 y no de la teoría clásica de que procede de las células de Merkel de la epidermis.
El CCM se presenta hasta en un 15% de los casos en colisión con otro tumor42. El más frecuente es el carcinoma de células escamosas53, y también se ha reportado la asociación con queratosis actínica, enfermedad de Bowen73 y carcinoma basocelular70. De manera anecdótica se ha descrito la asociación con quiste epidérmico, quiste tricolémico, tricoblastoma, poroma74 y lentigo maligno.
En ocasiones se ha descrito la regresión espontánea del tumor, pero el mecanismo por el que esto ocurre es desconocido. Takenaka et al.75 han sugerido un potencial efecto al realizar la biopsia mediante la estimulación del sistema inmune. Histológicamente la zona de regresión muestra infiltrado linfocitario perivascular, fundamentalmente de fenotipo CD8 y fibrosis. Se ha sugerido que los mecanismos de defensa inmunológica del huésped son determinantes en el proceso del CCM, lo cual abre la posibilidad de aplicación de terapia inmunológica en el futuro.
En la tabla 1 se detallan aquellos aspectos histológicos que deberían estar reflejados en el informe anatomopatológico ante un diagnóstico de CCM76–78.
Datos anatomopatológicos que debe incluir un informe de diagnóstico histológico de carcinoma de células de Merkel
| Tumor |
| Tipo de procedimiento (biopsia, exéresis; reexéresis, otra:______) |
| Tumor macroscópico (presente o ausente) |
| Localización tumoral:___________ |
| Tamaño tumoral (cm):___________ |
| Grosor tumoral (mm)a:____________ |
| Márgenes (laterales y profundos/afectados o libres) (distancia______mm) |
| Invasión linfovascular (presente o ausente) |
| Invasión de músculo, hueso, fascia o cartílago |
| Índice mitótico (por mm2)a:______/mm2 |
| Infiltrado linfocitario peritumorala (presencia o ausencia) (denso o no denso) (tipo celular de infiltrado: linfocitario, plasmocitario, otro:_____) |
| Infiltrado linfocitario intratumorala (presencia o ausencia) (denso o no denso) (tipo celular de infiltrado: linfocitario, plasmocitario, otro:_____) |
| Patrón de crecimiento tumorala (nodular o infiltrativo) |
| Presencia de diferenciación divergentea: escamosa, ecrina, melanocítica, sarcomatosa, otra:_______) |
| Presencia de otro tumor asociadoa (carcinoma escamoso, carcinoma basocelular, queratosis actínica, otro:___________) |
| Técnicas inmunohistoquímicas: |
| CKAE1/AE3 (positivo [____%], negativo) |
| CK20 (positivo [____%], negativo) |
| CK7 (positivo [____%], negativo) |
| Cromogranina (positivo [____%], negativo) |
| Sinaptofisina (positivo [____%], negativo) |
| CD56 (positivo [____%], negativo) |
| TTF1 (positivo [____%], negativo) |
| Ganglio |
| Tipo de procedimiento (ganglio centinela, linfadenectomía) |
| Localización |
| Diagnóstico con hematoxilina: positivo, negativo, equívoco |
| Diagnóstico con inmunohistoquímica: positivo, negativo, equívoco |
| Técnica inmunohistoquímica empleada: ____________ |
| Número de ganglios (total):______ y número de ganglios afectos_____ |
| Carga tumoral (porcentaje área afecta y tamaño del foco metastásico más grande____mm) |
| Localización de la metástasis: subcapsular, parénquima, centro germinal |
| Extensión extranodal (presente, ausente, equívoco) |
| Estadificación patológica (pTNM):____________ |
El estudio inmunohistoquímico ayuda en el diagnóstico de CCM, porque se trata de un carcinoma neuroendocrino, y presenta una expresión conjunta de citoqueratinas y marcadores neuroendocrinos. Se ha descrito un riesgo de error en el diagnóstico de alrededor del 66% si se utiliza solo la microscopia óptica79.
La mayoría de los CCM se marcan positivamente para las citoqueratinas de bajo peso molecular (CK8, 18, 19 y 20), mientras que las citoqueratinas de alto peso molecular (CK7) no se expresan. En la mayoría de los estudios publicados consideran muy útil valorar la expresión de la CK20 y la CK780, que es positiva en la primera y negativa en la segunda en el 90% de los casos. La CK20 presenta una tinción citoplasmática paranuclear, en motas, muy característica (dot-like) (fig. 4), y es uno de los principales pilares para su diagnóstico. Sin embargo, hay que señalar que la ausencia de positividad para CK20 no descarta el diagnóstico de CCM. Incluso se han descrito casos que presentan positivad para CK7 y negatividad para CK2081.
. B: microscopia electrónica de una célula tumoral donde se aprecian todos los filamentos intermedios agrupados.")
Entre los marcadores neuroendocrinos la enolasa neuronal específica es muy sensible, pero poco específica, ya que aparece en otras neoplasias (melanoma, otros tumores neuroendocrinos y carcinoma de pulmón de células pequeñas). La cromogranina y la sinaptofisina son proteínas presentes en los gránulos neurosecretores (fig. 5), y expresan una tinción citoplasmática típica en cúmulos globulares del 100% cromogranina B, 72% cromogranina A y 50% sinaptofisina42,82,83. En este sentido cabe reseñar la inmunotinción para CD56, una glucoproteína presente en las células neuroendocrinas y cuya expresión citoplasmática se ha descrito en aproximadamente el 94% de los CCM84.
 con la tinción citoplasmática granular característica. B y C: microscopia electrónica que corresponde a los gránulos electrodensos dispersos en el citoplasma que presentan las células tumorales del CCM.")
Inmunohistoquímica del carcinoma de células de Merkel. A: tinción inmunohistoquímica de diferenciación neuroendocrina (cromogranina) con la tinción citoplasmática granular característica. B y C: microscopia electrónica que corresponde a los gránulos electrodensos dispersos en el citoplasma que presentan las células tumorales del CCM.
El diagnóstico diferencial histológico se debe realizar con otros tumores de células redondas y pequeñas, principalmente con metástasis cutánea de carcinoma de pulmón de células pequeñas, carcinoma epidermoide poco diferenciado, un linfoma o un melanoma anaplásico de células redondas. En la tabla 2 se recogen los principales diagnósticos diferenciales y las principales técnicas inmunohistoquímicas utilizadas. El diagnóstico definitivo de CCM requiere negatividad para la proteína S-100, antígeno común leucocitario y citoqueratinas de alto peso molecular85,86.
Diagnóstico diferencial inmunohistoquímico del carcinoma de células de Merkel
| Tumor | CK | CK20 | CK7 | TTF-1/MASH1 | CD56/Cr | LCA | S-100 | CD99 |
|---|---|---|---|---|---|---|---|---|
| Carcinoma de Merkel | + | + | - | - | + | - | - | +/- |
| Carcinoma de células pequeñas de pulmón | + | - | + | + | + | - | - | +/- |
| Linfoma | - | - | - | - | - | + | - | - |
| Melanona | -a | - | - | - | + | - | + | - |
| Sarcoma de Ewing | -a | - | - | - | + | - | - | + |
+/-: el CCM y el carcinoma de células pequeñas de pulmón expresan CD99 a nivel citoplasmático pero no de membrana; CK: citoqueratinas (AE1/AE3); Cr: cromogranina; LCA: antígeno leucocitario común; MASH1: en humanos el gen que codifica es ASCL1 (mammalian achaete-scute hololog); TTF-1: factor de transcripción del tiroides.
La tinción inmunohistoquímica de membrana del CD99 es característica del sarcoma de Ewing. Sin embargo, en varias series42,87 un 40-55% de los CCM eran positivos para CD99, aunque a nivel citoplásmico.
El factor de trascripción tiroidea (TTF-1) es una proteína nuclear que interviene en la activación de la trascripción durante la embriogénesis en el tiroides y el epitelio respiratorio88. Es típicamente positivo en el cáncer de pulmón y tiroides y negativo en el CCM, de modo que se considera más específica del CCM la negatividad del TTF-1 que la positividad para CK20, ya que la CK20 puede ser negativa hasta en un 20% de los casos, mientras que el CCM prácticamente nunca expresa el TTF-1 (3%)89–91. Estudios recientes han encontrado la utilidad del MASH1, un gen relacionado con el desarrollo embriológico de células del cerebro y del sistema neuroendocrino, en el diagnóstico diferencial entre las metástasis cutáneas por un carcinoma de células pequeñas del pulmón y el CCM, sobre todo en casos raros positivos para TTF-1. El CCM no expresa MASH1, mientras que el 83% de carcinomas de células pequeñas del pulmón resultan positivos para este marcador91,92. De este modo, una combinación de TTF-1, MASH1 y CK 20 debería proporcionar la mayor sensibilidad y especificidad para distinguir entre CCM y otros carcinomas de células pequeñas.
En resumen, la detección inmunohistoquímica de filamentos intermedios, fundamentalmente CK2049 y marcadores neuroendocrinos como CD56, cromogranina o sinaptofisina, y la ausencia de expresión de TTF1, MASH1, CK7, proteína S-100 y antígeno leucocitario común (ALC) es útil en diferenciar CCM de otros tumores de células redondas y azules de la piel (tabla 2)43.
Estudio ultraestructuralLa microscopia electrónica ha tenido un papel clave en el conocimiento de este tumor93, pero en la actualidad no se solicita, puesto que la microscopia óptica y la inmunohistoquímica son suficientes para el diagnóstico. Las células del CCM presentan las mismas características que las células de Merkel, un núcleo lobulado y múltiples pequeños nucléolos. Dentro del citoplasma se aprecian abundantes ribosomas y un aparato de Golgi prominente.
Dos elementos ultraestructurales son característicos de la célula del CCM: los filamentos intermedios (fig. 4) y los gránulos electrodensos94 (fig. 5). Característicamente los gránulos electrodensos intracitoplasmáticos son de entre 100 y 250nm de diámetro. El carcinoma de células pequeñas de pulmón y otros carcinomas del sistema neuroendocrino también presentan gránulos electrodensos.
Alrededor del citoplasma de las células neoplásicas también se aprecian agregados paranucleares de filamentos intermedios o cuerpos fibrosos. Los filamentos intermedios se disponen en el carcinoma de células pequeñas de pulmón en una posición perinuclear, a diferencia del CCM que es típicamente paranuclear.
Estudio citogenético y molecularExisten alteraciones citogenéticas en un 30-47% de los CCM, fundamentalmente en el cromosoma 1 y también en los cromosomas 3, 11 y 1395. Estudios de hibridación genómica comparada encuentran ganancias y pérdidas muy similares a las observadas en el cáncer de pulmón de células pequeñas.
Se han descrito alteraciones en el oncogén bcl-296 y mutaciones del gen P5397, que podrían estar en relación con la progresión y una mayor agresividad del tumor. Parece ser que el número de mutaciones somáticas en el CCM está inversamente relacionado con la presencia del virus de polioma. Las muestras poliomavirus positivas presentan pocas mutaciones y las muestras poliomavirus negativas tienen una gran cantidad de mutaciones y una firma mutacional (RB y P53) que se corresponde con las mutaciones observadas en el cáncer de células pequeñas de pulmón98,99.
EstadificaciónDeterminar el estadio de los pacientes con CCM tiene gran importancia para establecer el pronóstico y tratamiento más adecuado. Clásicamente la estadificación se realizaba de manera sencilla según el sistema descrito por Yiengpruksawan41: estadio i enfermedad localizada en la piel (IA-lesión<2cm; IB-lesión≥2cm); estadio ii enfermedad ganglionar regional; estadio III enfermedad metastásica. En el año 2010 la American Joint Committee on Cancer propone una nueva estadificación más completa100 (tabla 3), basada en el análisis de más de 4.000 casos. En ella se pone de manifiesto la importancia del tamaño tumoral (T) y el conocimiento del estado de los ganglios linfáticos regionales (N) y metástasis (M) en relación con la supervivencia. Según esta clasificación existen 4 estadios clínicos en el momento del diagnóstico (tabla 4). Los primeros 2 estadios corresponden a pacientes con la enfermedad localizada únicamente en la piel, el tercer estadio incluye pacientes con afectación de los ganglios regionales, y el cuarto estadio refleja diseminación metastásica. Los pacientes con verdadera enfermedad local (estadio i-ii) tienen alrededor del 50% de probabilidades de sobrevivir a los 5 años, porcentaje que decrece a menos del 20% cuando existe enfermedad metastásica (estadio iv) (tabla 4). El tamaño tumoral es el factor pronóstico más importante cuando el tumor está localizado solo en la piel (estadio i y ii). La mayoría de estudios encuentran que los CCM con un tamaño superior a 2cm presentan peor pronóstico (≥T2)77. Sin embargo, no se ha visto que existan diferencias significativas entre un tamaño >2cm-≤5 (T2) y >5cm (T3) y, por eso, ambos se incluyen en el mismo estadio ii (tabla 4). No obstante, independientemente del tamaño tumoral, si la lesión de la piel invade la fascia, el músculo, el hueso o el cartílago pasa a considerarse un T4 y sí que implica un peor pronóstico.
TNM según la American Joint Committee on Cancer para el carcinoma de células de Merkel
| T | N | M |
|---|---|---|
| Tx, el tumor primario no puede ser evaluado | Nx, La afectación ganglionar no puede ser evaluada | Mx, las metástasis no pueden ser evaluadas |
| T0, no hay evidencia de tumor primario | N0, no hay afectación ganglionar | M0, no hay metástasis |
| Tis, tumor primario in situ | -cN0, no hay afectación ganglionar detectada clínicamente (inspección, palpación y/o radiología) | M1, metástasis a distancia |
| T1, tumor primario ≤2cm | -pN0, no hay afectación ganglionar detectada tras estudio anatomopatológico | -M1a, metástasis a la piel, tejido celular subcutáneo o ganglios linfáticos a distancia |
| T2, tumor primario >2-≤5cm | -pNx, ganglios no estudiados histológicamente | -M1b, metástasis al pulmón |
| T3, tumor primario >5cm | N1a, micrometástasisa | -M1c, metástasis en otras vísceras |
| T4, tumor primario afectando el hueso, el músculo, la fascia o el cartílago | N1b, macrometástasisb | |
| N2, metástasis en tránsitoc |
Micrometástasis: afectación del ganglio clínicamente oculta y que se detecta en el ganglio centinela o en una linfadenectomía electiva.
Macrometástasis: existe evidencia clínica y comprobación anatomopatológica del ganglio linfático regional o en punción biopsia.
Metástasis en tránsito: metástasis que se localiza entre el tumor primario y los ganglios linfáticos regionales o distalmente a la tumoración primaria.Fuente: Lemos et al.100
Estatificación según la American Joint Committee on Cancer para el carcinoma de células de Merkel
| Estadio | Tumor | Ganglios | Metástasis | Supervivencia a 5 años (%)a |
|---|---|---|---|---|
| 0 | Tis | N0 | M0 | 100 |
| IA | T1 | pN0 | M0 | 79 |
| IB | T1 | cN0 | M0 | 60 |
| IIA | T2/T3 | pN0 | M0 | 58 |
| IIB | T2/T3 | cN0 | M0 | 49 |
| IIC | T4 | N0 | M0 | 47 |
| IIIA | Cualquier T | N1a | M0 | 42 |
| IIIB | Cualquier T | N1b/N2 | M0 | 26 |
| IV | Cualquier T | Cualquier N | M1 | 18 |
En la nueva clasificación se distingue si la ausencia de afectación ganglionar está confirmada mediante estudio anatomopatológico (pN0) o no lo está (cN0) y su negatividad se establece solo por criterios clínicos y radiológicos. Esta distinción entre clínica e histología es muy importante, pues hasta un tercio de los pacientes con aparente enfermedad local presentan metástasis ocultas101. No obstante, la presencia de micrometástasis (N1a) implica un pronóstico mucho mejor (42% de supervivencia a los 5 años) que las macrometástasis (N1b) (26% de supervivencia a los 5 años) (tabla 4).
Los pacientes que comienzan con enfermedad metastásica al diagnóstico presentan una supervivencia aproximada del 44% al año100. La nueva clasificación realiza categorías en función del lugar de la metástasis (tablas 3 y 4), sin embargo la localización de la metástasis no diferencia la supervivencia.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


