






INTRODUCCIÓN
El xantogranuloma es una lesión dermatológica caracterizada histológicamente por una proliferación de histiocitos. Es de carácter benigno y tiende a la regresión espontánea. La variedad clínica más frecuente afecta a niños pequeños y se presenta como una o múltiples lesiones papulosas o nódulos amarillo-par-duzcos distribuidos preferentemente en la parte alta del cuerpo, especialmente en cabeza y cuello, entidad denominada xantogranuloma juvenil (XGJ). Es la histiocitosis de células no Langerhans (HCNL) más frecuente. Su aparición en el adulto es más rara y desde que se describió por primera vez en 1963 por Gartmann y Tritsch1 , se han publicado pocos casos en la literatura médica1-12 . Por lo general se presenta como una lesión única, y es excepcional su presentación con lesiones múltiples diseminadas. Esta variante aparece publicada en la literatura médica con el nombre de xantogranulomas múltiples del adulto (XGMA)1-7 .
DESCRIPCIÓN DEL CASO
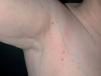
Una mujer de 53 años sin antecedentes medicoquirúrgicos de interés fue vista por primera vez en nuestras consultas en junio de 2001. Consultaba por la aparición progresiva, desde hacía 7 años, de lesiones múltiples, de forma generalizada por su superficie corporal. En la exploración se apreciaban múltiples pápulas de coloración parduzco-amarillentas, de consistencia firme, en número aproximado de 100, distribuidas simétricamente a ambos lados del tronco, abdomen y zona proximal de extremidades (fig. 1). Las lesiones eran, por lo general, de pocos milímetros, aunque algunas de ellas alcanzaban 1 cm de diámetro (fig. 2). La paciente no refería prurito ni otra sintomatología acompañante y su estado general era bueno. Tampoco se apreció afectación de mucosas.
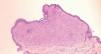
Se realizó estudio histológico de varias lesiones, mostrando todas ellas los mismos hallazgos anatomopatológicos; una densa reacción dérmica inflamatoria linfohistiocitaria (fig. 3), junto con un infiltrado de células xantomatosas de aspecto espumoso y gran número de linfocitos y células gigantes multinucleadas tipo reacción a cuerpo extraño y tipo Touton (fig. 4). Las técnicas de inmunohistoquímica realizadas resultaron positivas para marcadores de células de estirpe monocito-macrófago (CD68) y de dendrocitos dérmicos (FXIIIa), y fueron negativas para células de Langerhans (S100 y CD1a). La analítica general, incluido perfil lipídico y estudio hematológico, se encontraba dentro del rango de la normalidad, así como el estudio de extensión realizado para descartar afectación visceral (serie ósea, ecografía abdominal y tomografía computarizada [TC] toracoabdominopélvica). En la exploración oftalmológica no se detectó compromiso ocular asociado.

Fig. 1.—Lesiones papulosas de pocos milímetros, amarillo-parduzcas, distribuidas en tronco y zona proximal de extremidades.
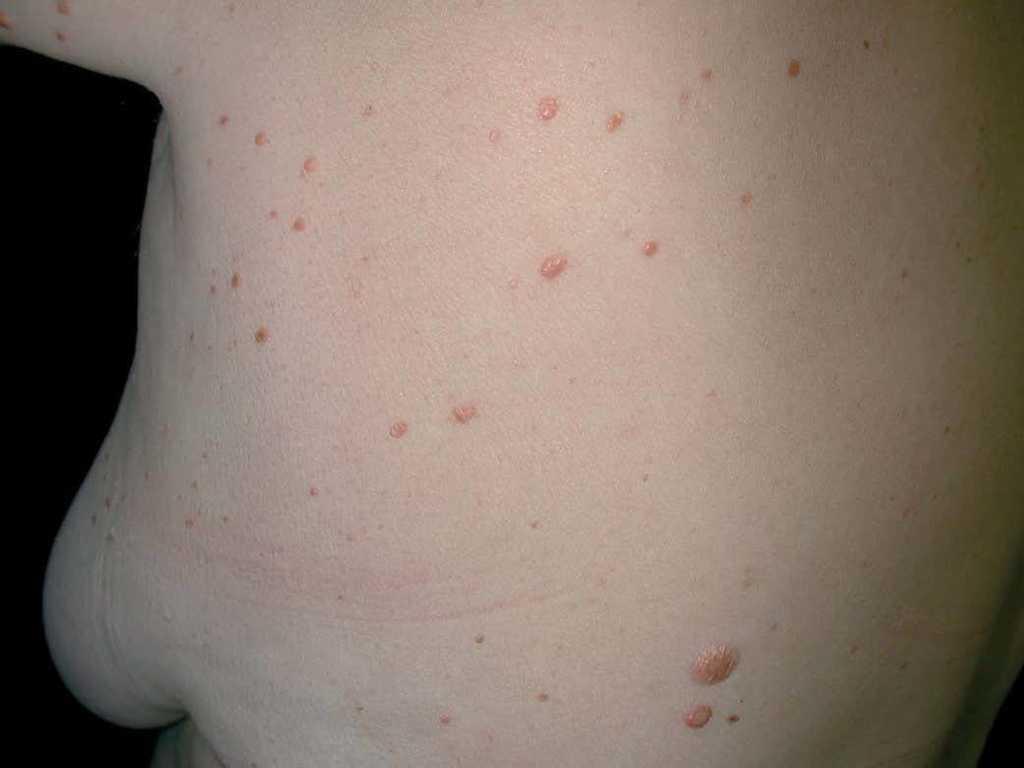
Fig. 2.—Múltiples lesiones papulonodulares de consistencia firme en tronco.
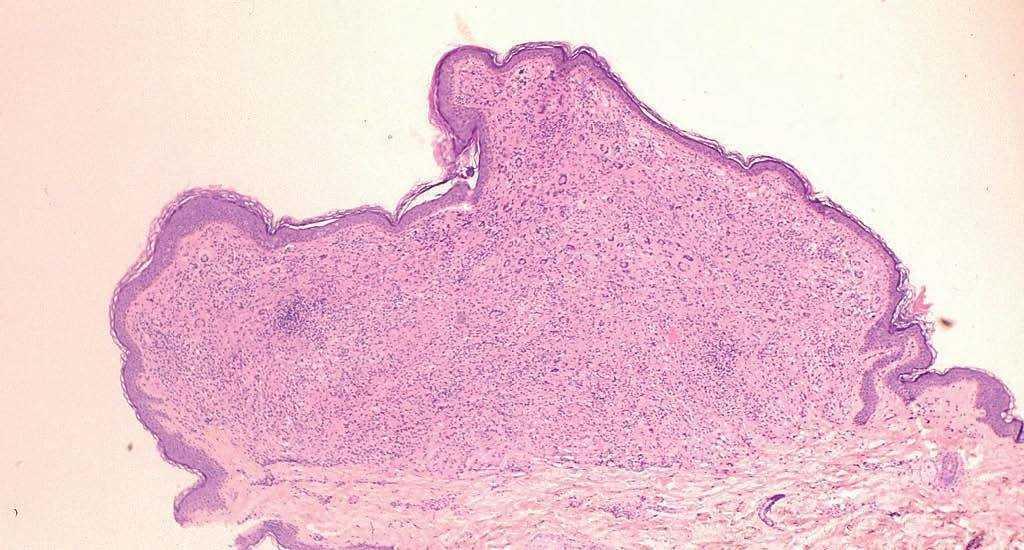
Fig. 3.—Densa reacción inflamatoria linfohistiocitaria bien delimitada que ocupa toda la dermis papilar. (Hematoxilina-eosina, ×40.)
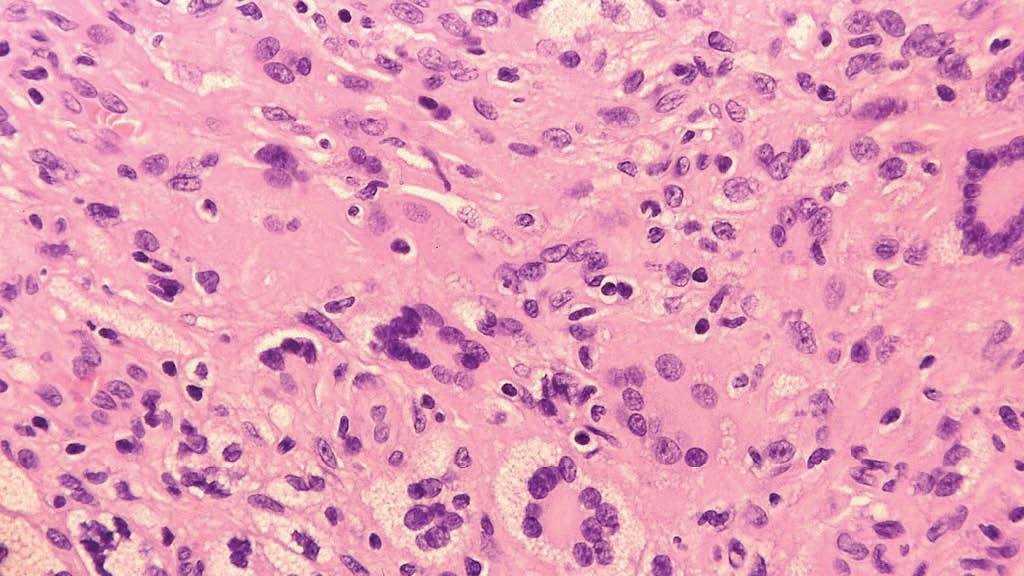
Fig. 4.—Infiltrado constituido principalmente por células histiocitarias y células xantomatosas de aspecto espumoso, junto con gran número de linfocitos y células gigantes multinucleadas tipo Touton. (Hematoxilina-eosina, ×400.)

TABLA 1. CLASIFICACIÓN DE LAS HISTIOCITOSIS
Durante el último año de seguimiento han ido apareciendo nuevas lesiones, algunas incluso en cara, mientras que otras han ido involucionando de manera espontánea dejando zonas de atrofia residual, pudiéndose observar lesiones en diferente estadio evolutivo. Con el diagnóstico de xantrogranulomas múltiples del adulto, se inició tratamiento con crioterapia con nitrógeno líquido en algunas lesiones, observándose una eficacia moderada; las pápulas se aplanaban y algunas incluso llegaban a resolverse, pero dejaban un área de atrofia residual. En la actualidad, y tras 7 años del inicio del cuadro, la paciente sigue en observación realizando controles periódicos tanto clínicos como hematológicos.
DISCUSIÓN
Las histiocitosis son procesos reactivos, en ocasiones malignos, que se caracterizan por la proliferación de histiocitos, células de la línea monocito-macrófago, que infiltran la piel y pueden afectar también a otros órganos y tejidos (tabla 1). Las histiocitosis no X o HCNL, como se prefiere llamar en la actualidad, comprenden un amplio espectro de enfermedades que en muchas ocasiones pueden resultar difíciles de diferenciar entre sí, ya que comparten rasgos clínicos e histológicos. Se caracterizan por la proliferación de histiocitos que no contienen gránulos de Birbeck en su citoplasma. Se han intentado múltiples clasificaciones que pretenden aclarar este grupo tan confuso de enfermedades 13-15 . En 1996 se propuso un concepto unificador que consiste en clasificar las diferentes entidades dependiendo del tipo de célula histiocitaria que predomine 14 . Otros autores proponen que las HCNL representan en realidad un espectro de una misma enfermedad en las que el dendrocito dérmico muestra diferentes niveles de maduración 15 . Así, el XGJ quedaría en el extremo correspondiente a las células más inmaduras.
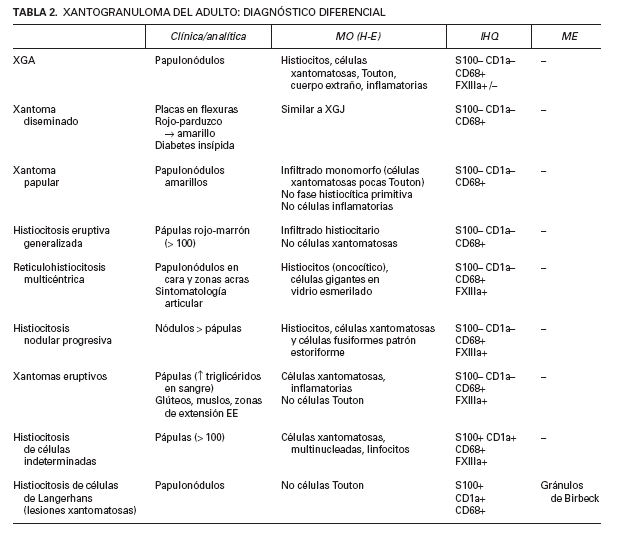
TABLA 2. XANTOGRANULOMA DEL ADULTO: DIAGNÓSTICO DIFERENCIAL
El XGJ se considera la forma más frecuente de HCNL. En la mayoría de los casos se manifiesta en el primer año de vida, aunque puede hacerlo más tarde. Consiste en la presencia de una única o, más frecuentemente, numerosas lesiones papulonodulares, de coloración rojo-parduzca que van adquiriendo un color amarillento, distribuidas principalmente en cabeza y cuello. La evolución es hacia la resolución espontánea en 3 a 6 años. Su hallazgo en un adulto es mucho más rara.
El xantogranuloma del adulto (XGA) fue descrito por primera vez por Gartmann y Tritsch en 19631 . Se han publicado unos 120 casos desde entonces en la literatura médica, sobre todo en la japonesa. Aparece casi siempre como lesión única, siendo mucho más rara su presentación como lesiones múltiples, que se denomina XGMA. Se caracteriza por la presencia de numerosas lesiones sobreelevadas, de pocos milímetros a varios centímetros de diámetro, de consistencia firme, color amarillo-parduzco y distribuidas fundamentalmente por el tronco y extremidades. Las lesiones son asintomáticas y, a diferencia de la forma juvenil, aunque pueden resolverse de forma espontánea, suelen hacerlo incluso hasta 15 años después del diagnóstico. En ocasiones, la forma juvenil se asocia a manifestaciones extracutáneas, de las que la más frecuente es la afectación ocular con compromiso uveal y riesgo de hemorragia y glaucoma. También puede ir asociado a compromiso de otros órganos e incluso están descritos casos asociados a síndromes mieloproliferativos crónicos. La afectación extracutánea en el adulto es excepcional y, hasta el momento sólo hay publicados 2 casos con evolución a leucemia linfoide crónica y gammapatía monoclonal, respectivamente5 , un caso descrito asociado a trombocitosis esencial6 y un paciente de 25 años diagnosticado de trisomía 21 y leucemia linfoide aguda que presentó un cuadro de xantogranulomas eruptivos durante una reactivación de su leucemia7 . Por otro lado, hay documentados casos de xantogranulomas oculares en adultos acompañando o no a otras manifestaciones cutáneas8 .
El estudio histológico de las lesiones es característico, con hallazgos similares a los encontrados en la forma juvenil y con una inmunohistoquímica que permite diferenciarlo sin problemas de las formas más agresivas de histiocitosis de células de Langerhans (HCL)9,10 . Consiste en la presencia de un infiltrado denso linfohistiocitario bien circunscrito en dermis papilar, que puede ocupar la totalidad de la dermis. En las fases más tempranas se observa un infiltrado histiocítico monomorfo, sin lípidos. En fases más tardías, cuando las lesiones están más evolucionadas, se aprecian en el infiltrado células espumosas, que corresponden a histiocitos cargados de lípidos, junto con células gigantes tipo Touton y tipo reactivas a cuerpo extraño. Acompañando a este infiltrado hay también cantidades variables de linfocitos, eosinófilos y neutrófilos. En las formas más avanzadas puede haber fibrosis. En el estudio inmunohistoquímico, los marcadores de células macrofágicas, CD68 y HAM56 son positivos, así como el marcador de dendrocitos dérmicos FXIIIa, mientras que los marcadores de células de Langerhans S100 y CD1a resultan negativos. En el estudio ultraestructural no se encuentran gránulos de Birbeck en el interior de las células.
El diagnóstico diferencial comprende las diferentes entidades englobadas dentro de las HCNL, así como las HCL que deben descartarse en primer lugar por tratarse de enfermedades eventualmente más agresivas y con manifestaciones sistémicas asociadas (tabla 2).
En cuanto al manejo de este cuadro, la mayoría de los autores prefieren la observación del mismo aunque se ha propuesto el tratamiento con crioterapia local, láser de dióxido de carbono (CO2), e incluso extirpación simple de las lesiones más grandes.
Las manifestaciones extracutáneas en estos casos son excepcionales. Aun así, e independientemente de la decisión terapéutica tomada, se debe vigilar la evolución del paciente, realizando controles hematológicos periódicos, por haberse descrito algún caso asociado a discrasias hematológicas5-7 .
Correspondencia:
Belén Navajas. Servicio de Dermatología. Hospital de Cruces. Pl. de Cruces, s/n. 48903 Barakaldo. Vizcaya. España. bnavajas@aedv.es
Recibido el 25 de agosto de 2004. Aceptado el 17 de noviembre de 2004.


