



INTRODUCCION
La presencia de úlceras en pacientes con dermatomiositis ocurre habitualmente en la variante juvenil en asociación con calcinosis cutánea1,2, cuya aparición se atribuye a una vasculitis2 o una vasculopatía1. Sin embargo, en pacientes adultos esta asociación es excepcional y se relaciona con una enfermedad más agresiva o una neoplasia interna2. El mecanismo patogénico de las úlceras en estos casos está menos estudiado que en niños, y probablemente no se deban a una vasculitis, ya que en los casos en que se realizó un estudio histopatológico se descartó la existencia de ésta2-4.
Presentamos 2 casos de pacientes afectados de dermatomiositis que tenían lesiones ulcerosas ampliamente distribuidas por el tronco y las extremidades superiores. En el estudio histopatológico de las lesiones se hallaron alteraciones de los vasos en forma de trombos y dilatación con presencia de material hialino en la pared. Creemos que el origen de las úlceras en nuestros casos se debe a una vasculopatía livedoide y pensamos que ésta pudiera ser sea la causa de las lesiones ulcerosas en los pacientes adultos afectados de dermatomiositis.
DESCRIPCION DE LOS CASOS
Caso 1
Un varón de 42 años, con antecedentes de tuberculosis y dislipemia, fue diagnosticado de dermatomiositis en 1991, por la que recibió tratamiento con dosis variables de corticoides y metotrexato hasta su fallecimiento por sepsis en 2000. Los estudios complementarios realizados descartaron la existencia de una neoplasia o de un síndrome antifosfolípido asociados, pero se descubrieron un seudoquiste de cabeza de páncreas y un hígado graso en la tomografía computarizada y en la ecografía abdominal, y una anemia interpretada como de enfermedad crónica en el hemograma. El servicio de dermatología fue requerido para valorar unas lesiones que estaban presentes desde el inicio de su dermatomiositis, seguían un curso paralelo a ésta, y mejoraban al aumentar la dosis de corticoides por vía sistémica. Se trataba de úlceras dolorosas que se distribuían sobre todo por extremidades superiores y áreas periaxilares, incluyendo el dorso de las manos. Estas úlceras curaban dejando una cicatriz blanco-marfileña de aspecto atrófico.
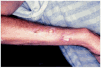
En la exploración se observaban entre 10 y 20 úlceras de 1 cm de diámetro aproximadamente, distribuidas por la zona de extensión de las extremidades superiores (fig. 1) y zona periaxilar, de fondo sucio y borde eritematoso sobreelevado. Junto a este tipo de lesiones presentaba otras en forma de máculas redondeadas de borde irregular, cuyo centro era blanco-marfileño y el borde estaba constituido por un halo eritematoso formado por telangiectasias. Además, se hallaron otras alteraciones cutáneas como livedo reticularis en flancos y extremidades superiores sobre todo, unos dedos afilados y algo esclerosos y una piel de aspecto poiquilodérmico en dorso de manos, espalda y área del escote. No se apreció eritema heliotropo en ese momento, aunque lo había presentado con anterioridad, y era evidente una facies cushingoide.
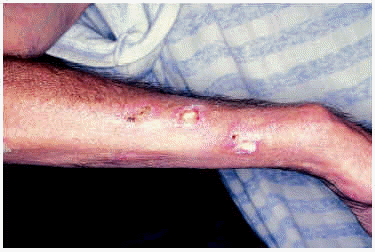
Fig. 1.--Caso 1. Úlceras redondeadas de fondo sucio y borde sobre-elevado.
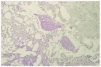
Un cultivo del exudado de una úlcera aisló Staphylococcus aureus, aunque el tratamiento antibiótico no modificó la evolución del cuadro. El estudio histopatológico de una lesión ulcerosa de reciente aparición, localizada en brazo, mostró una alteración en cuña con una úlcera superficial acompañada de un denso infiltrado inflamatorio difuso en el área superficial constituido por linfocitos, histiocitos y neutrófilos. En el vértice de la cuña, situado en la unión dermohipodérmica, se apreciaba una arteriola que contenía un trombo de fibrina que ocluía por completo la luz (fig. 2). A ese nivel se observaban escasas células inflamatorias sin relación con el vaso. Alrededor de la úlcera, se observaban diversas alteraciones en los vasos sanguíneos. Algunos vasos de la dermis mostraban una luz dilatada con una pared engrosada que contenía un material hialino (fig. 3), mientras que otros presentaban una luz estrecha con una pared rodeada de fibrina, muchos de ellos con trombos intraluminales.
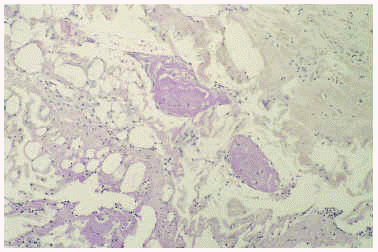
Fig. 2.--Caso 1. Arteriola localizada en el unión dermoepidérmica, en el ápex de la úlcera. En este punto se observa escaso infiltrado inflamatorio que no estaba en relación con el vaso; sin embargo, se aprecia un trombo de fibrina en su luz.

Fig 3.--Caso 1. Vaso dilatado con un material hialino en la pared. Este vaso estaba localizado en la dermis papilar, por fuera de la úlcera con una epidermis suprayacente normal.
Caso 2
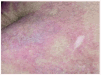
Una mujer de 58 años, alérgica a la penicilina, con antecedentes de hernia discal, bocio, apendicectomía y ooforectomía derecha, diagnosticada en el año 2000 de dermatomiositis amiopática por presentar telangiectasias periungueales, pápulas de Gottron, placas poiquilodérmicas en glúteos, y de pápulas eritematoedematosas faciales, con empeoramiento tras la exposición solar. Los estudios realizados descartaron la presencia de un síndrome antifosfolípido o una neoplasia asociada, aunque se descubrieron un quiste en el riñón derecho mediante una ecografía abdominal y una gastroduodenitis erosiva mediante una endoscopia. Al año del inicio de la dermatomiositis, la paciente desarrolló ocho lesiones ulcerosas intensamente dolorosas que se distribuían por brazos, dedos y dorso de manos, glúteos y muslos. Tras aumentar la dosis de prednisona a 60 mg/día (1 mg/kg/día) y aplicar ácido fusídico de forma tópica, las lesiones curaron dejando unas cicatrices blanco-marfileñas, algunas con telangiectasias en su interior (fig. 4).

Fig 4.--Caso 2. Cicatriz blanco-marfileña de aspecto atrófico en un área de poiquilodermia, evolución de una úlcera previa.
Se realizaron dos biopsias de las lesiones ulcerosas, una del muslo y otra del brazo, que mostraron hallazgos histopatológicos similares. Se observaba una úlcera superficial en la que se apreciaba una costra que contenía fibrina, bacterias y un infiltrado inflamatorio. Por debajo de ésta se apreciaban vasos con trombos de fibrina intraluminales, sin presencia de infiltrado inflamatorio perivascular. En la periferia de la úlcera y por debajo de una epidermis normal, se apreciaba vasodilatación y engrosamiento con hialinización de las paredes vasculares. En esa zona no se observaban células inflamatorias o bien eran muy escasas.
COMENTARIO
Existe gran confusión en torno al término atrofia blanca5. Esta entidad, descrita inicialmente por Milian en 19296, debe su nomenclatura al aspecto de las lesiones residuales en forma de cicatrices de forma irregular, coloración blanco-marfileña, que contienen un moteado telangiectásico en su superficie. Desde entonces se ha denominado de muchas maneras: capilaritis alba, livedo reticularis con ulceraciones en verano o livedo reticularis con ulceraciones en invierno, según la estación en que predominaran, vasculitis livedoide, en la creencia de un origen vasculítico del proceso, vasculitis segmentaria hialinizante, indicando algunos de sus hallazgos histopatológicos, vasculitis de la atrofia blanca7 y, de forma más reciente, PURPLE (painful Purpuric Ulcers with Reticular Patterning of the Lower Extremities)8 y vasculopatía livedoide9. Además de la nomenclatura, el hecho que estas lesiones residuales puedan ser secundarias a otras enfermedades sin relación con la vasculopatía livedoide, como estasis venosa, o una vasculitis leucocitoclástica ha aumentado la confusión sobre esta entidad5,10.
Actualmente la teoría patogénica más aceptada explica las lesiones de vasculopatía livedoide como secundarias a una oclusión vascular debida a una alteración en el proceso de trombosis-fibrinolisis, sin intervención de células inflamatorias5,9,11. Las alteraciones halladas no son constantes en todos los pacientes, por lo que probablemente la vasculopatía livedoide sea un síndrome en el que esta alteración del proceso trombosis-fibrinolisis pueda deberse a un gran número de causas8,10. De este modo se explica la elevada incidencia de síndrome antifosfolípido en este grupo de pacientes12, o la asociación a conectivopatías8,10. De forma ocasional se han encontrado la presencia de una mutación en el factor V de Leyden13, una alteración en la agregación plaquetaria14 o una crioglobulinemia11, entre otras alteraciones. Probablemente estas diferencias patogénicas expliquen las respuestas dispares a los tratamientos, antiagregantes (aspirina, dipiridamol), mientras que otros pacientes precisan fibrinolíticos (etilenestrenol, fenformina, activador del plasminógeno tisular) y aún otros responden a vasodilatadores (nifedipino, sulfasalazina), fármacos que disminuyen la viscosidad sanguínea (pentoxifilina) o a anticoagulantes (warfarina, heparina)10.
En cuanto a la sintomatología, inicialmente se manifiesta como petequias o pápulas purpúricas que se necrosan formando úlceras dolorosas de contornos angulares y evolución tórpida, que al curar dejan una característica cicatriz estrellada blanco-marfileña salpicada por telangiectasias, de bordes hiperpigmentados, con o sin livedo alrededor8. En la inmensa mayoría de los casos estas lesiones se localizan en las piernas, principalmente en la zona del tobillo10. No obstante, y en especial cuando se asocia a conectivopatía, pueden presentar una sintomatología distinta, con una distribución más amplia y úlceras más pequeñas y numerosas. El diagnóstico en estos casos es más difícil de establecer y debe realizarse el diagnóstico diferencial con la enfermedad de Degos. Al respecto, el artículo de Black et al15 publicado en 1976 describe los casos de 2 pacientes con atrofia blanca asociada a lupus que presentaban cicatrices blancas atróficas semejantes a las producidas en la enfermedad de Degos. Previamente a este artículo se había publicado un caso de esclerodermia sistémica asociado a una «verdadera» enfermedad de Degos, ya que en la autopsia se encontraron lesiones en colon, riñón y páncreas16. En conclusión, el diagnóstico diferencial entre ambas enfermedades es obligado en los casos asociados a conectivopatías. Mientras para algunos autores este tipo de lesiones serían secundarias a la conectivopatía de base17,18, otros defienden que sería una variante de síndrome de Degos asociada a conectivopatía a la que denominan variante sintomática19. Nosotros, al igual que otros autores, incluyendo al propio Degos20, pensamos que estos pacientes presentan una vasculopatía livedoide15,20,21.
En cuanto a la dermatomiositis en adultos en particular, se han publicado pocos casos asociados a lesiones ulcerosas2-4,22-24. Salvo en el caso de Yamamoto et al25 en el que se encontró una vasculitis en el estudio histopatológico, en los casos en los que se realizó biopsia se descartó el origen vasculítico de las úlceras2-4, pero sin aportar una alternativa patogénica. Aunque en esos casos las descripciones histopatológicas no son muy precisas pensamos que podrían ser compatibles con una vasculopatía livedoide, ya que se describe la presencia de trombos4 o un material fibrinoide en la pared vascular3.
Stephansson et al26, al comparar pacientes con lupus eritematoso sistémico (LES) asociado a síndrome antifosfolípido en pacientes con LES sin síndrome antifosfolípido, encontraron lesiones semejantes a enfermedad de Degos en el 9 % (3/33) de los casos afectados por ambos procesos, lo que hace pensar que estos casos no son tan infrecuentes como la literatura especializada haría suponer. Por otro lado, este tipo de lesiones no están contempladas en revisiones de síndrome antifosfolípido, aunque sí la forma clásica de vasculopatía livedoide27.
El tratamiento de elección para este tipo de lesiones no está establecido. No se han ensayado los tratamientos habituales para la vasculopatía livedoide, y en la mayoría de los casos se instauró tratamiento corticoideo por vía sistémica. Nuestros casos y otros similares han respondido en mayor o menor medida a dosis altas de corticoides28. La inmunoglobulina por vía intravenosa supone una alternativa terapéutica con buenos resultados para los casos resistentes3,22.


