





INTRODUCCION
El reconocimiento de las manifestaciones clínicas precoces de las enfermedades hereditarias es importante, ya que permite la utilización de un tratamiento precoz, así como el consejo genético adecuado. En algunas de estas enfermedades es posible aplicar en el momento actual nuevas terapias basadas en la etiopatogenia del proceso, tal y como ocurre en la enfermedad de Fabry.
En ausencia de historia familiar conocida de enfermedad de Fabry, muchos casos no se descubren hasta la juventud (con una mediana de 29 años)1,2 cuando el proceso fisiopatológico de la enfermedad ya está avanzado. Además, con frecuencia la forma de presentación clínica es muy sutil, por lo que los síntomas son infravalorados o erróneamente atribuidos a otros diagnósticos como eritromelalgia, neurosis, síndrome de Raynaud, esclerosis múltiple, polineuropatía crónica intermitente desmielinizante, lupus, apendicitis agudas o incluso petequias2,3. Posteriormente, la sintomatología es mucho más consistente con implicación importante cardíaca, renal y del sistema nervioso central (SNC). Tradicionalmente la medicina sólo permitía realizar un tratamiento paliativo, que podía prolongar la vida del paciente pero que no actuaba sobre la causa de la enfermedad. Recientemente ha comenzado a utilizarse un tratamiento enzimático sustitutivo con resultados esperanzadores.
Se presenta un caso de enfermedad de Fabry que en la actualidad está siguiendo este tratamiento enzimático sustitutivo.
DESCRIPCION DEL CASO
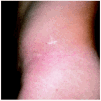
Un varón de 15 años consultó en el año 1997 por unas lesiones cutáneas acompañadas de dolor lancinante en los pulpejos de las manos y de los pies de varios minutos de duración después de practicar ejercicio físico. Los dolores remitían con el reposo. Como único antecedente familiar de interés, la madre presentaba una proteinuria crónica asociada a hipertensión arterial. La exploración física reveló la presencia de múltiples lesiones papulosas puntiformes, eritematosas, discretamente hiperqueratósicas, distribuidas por todo el tegumento, aunque con un predominio de las palmas, de las plantas y de los genitales (figs. 1 y 2). En la conjuntiva ocular llamaba la atención la presencia de una mayor tortuosidad de los vasos sanguíneos (fig. 3).
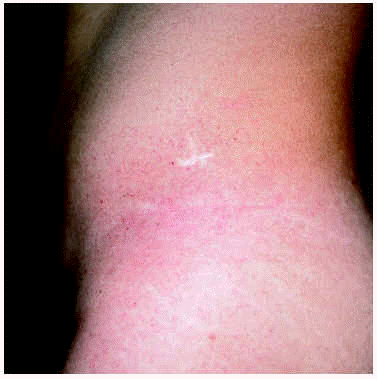
Fig. 1.--Máculas eritematosas algo hiperqueratósicas con distribución bilateral en flancos compatible con angioqueratomas corporis diffusum.
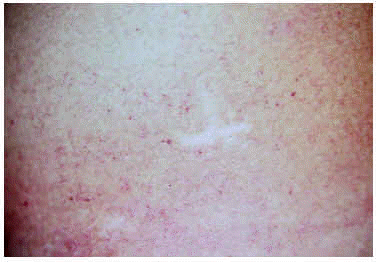
Fig. 2.--Detalle de las lesiones cutáneas.
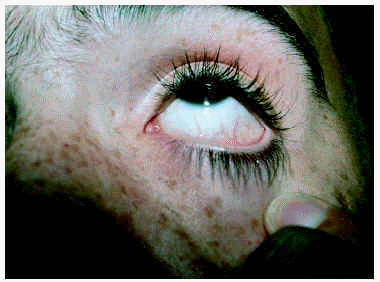
Fig. 3.--Vasos tortuosos en la conjuntiva ocular.
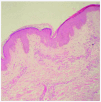
Se realizó una biopsia de una de las lesiones cutáneas cuyo estudio histológico mostró la presencia de vasos dilatados en el plexo vascular superficial de la dermis papilar. A mayor aumento se observaban abundantes vacuolas en el citoplasma de las células endoteliales (fig. 4). Todos estos hallazgos eran compatibles con angioqueratoma corporis diffusum. Se realizó un estudio analítico con hemograma y bioquímica, que incluyó las funciones renal y hepática, con valores dentro de los límites normales. Se evaluaron, además, los oligosacáridos en la orina que resultaron normales y se descartaron las glucoproteinosis (α y β -manosidosis, fucosidosis, aspartilglucosaminuria y sialidosis), gangliosidosis GM1 tipo 1, gangliosidosis GM2 tipo Sandhoff, galactosialidosis, enfermedad de Schindler y glucogenosis II, III y VI. El análisis enzimático en fibroblastos mostró unos valores normales para β -galactosidasa, hexosaminidasa y N-acetilneuraminidasa, con valores claramente disminuidos para α -galactosidasa (3,2 nmol/h/mg prot. frente a 56,5 del control), por lo que fue diagnosticado de enfermedad de Fabry. La madre del paciente presentaba unos valores en el rango de portadora (162 nmol/min/g de prot. frente a 1.144 del control). El análisis genético confirmó el estado de portadora y por lo tanto la heterozigosidad para esta enfermedad. Se practicó una biopsia renal cuyos hallazgos fueron compatibles a los referidos en la enfermedad de Fabry (fig. 5).
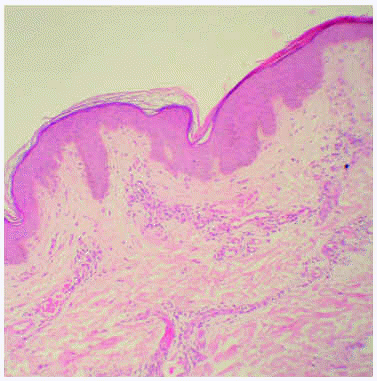
Fig. 4.--Detalle anatomopatológico de las lesiones cutáneas que muestra vasos dilatados en el plexo vascular superficial. (Hematoxilina-eosina, x20.)
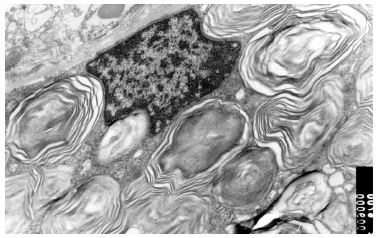
Fig. 5.--Imagen de microscopia electrónica de biopsia renal de la madre del paciente. Depósito de globotriaosilceramida renal.
Inicialmente sólo se pudo ofrecer al paciente un tratamiento paliativo del dolor neurálgico con analgésicos y antiepilépticos (fenitoína y carbamazepina) con un control pobre de la sintomatología. En febrero de 2001 se instauró la terapia de sustitución enzimática, tratamiento que sigue manteniendo en el momento actual. Recibe 0,2 mg/kg de peso de a -galactosidasa (Replagal®) en infusión de 40 min de duración que se realiza de manera bisemanal. Antes y después de la infusión se realizaron sendos controles de peso, de la presión arterial y de la frecuencia cardíaca, con objeto de monitorizar las reacciones transfusionales. No ha tenido ningún efecto secundario importante exceptuando una pérdida de aproximadamente 6 kg de peso. Posteriormente, también se inició el tratamiento de la madre del paciente. Con este tratamiento se obtuvo una disminución de las crisis dolorosas (valoradas de forma subjetiva por el paciente); lo que condujo a la supresión de los analgésicos de gran potencia que utilizaba (fenitoína y carbamazepina). No obstante, el paciente aún sufre crisis lancinantes dolorosas aunque en menor número y duración, que en la actualidad se controlan con analgésicos de menor potencia (metamizol y paracetamol).
Asimismo, disminuyeron en extensión las lesiones cutáneas. En lo referente a los niveles de globotriosilceramida plasmática, éstos siguieron un curso oscilante, aunque con tendencia a la disminución (7,5; 6,7; 5,1; 5; 4,7; 5,1; 4,3; 7,5) μmol/l (medición bimensual).
DISCUSION
La enfermedad de Fabry se debe a un error congénito del metabolismo glucofosfolipídico, consecuencia de un defecto en la actividad de la enzima lisosómica a -galactosidasa A (también llamada ceramida trihexosidasa). La enfermedad presenta una incidencia aproximada de 1 en 40.000-60.000 varones. El defecto enzimático está transmitido por un gen recesivo ligado al cromosoma X (localizado en Xq22). Aproximadamente se han recogido unas 150 mutaciones de este gen en la base de datos de mutaciones de los genes humanos3. Algunas de estas mutaciones se correlacionan con determinadas expresiones fenotípicas de la enfermedad. Este déficit enzimático conduce al depósito progresivo de glucoesfingolípidos neutros no hidrolizados con fragmentos terminales α -galactosil, procedentes del recambio celular continuo de la membrana celular3-8. En consecuencia, a lo largo de décadas, se acumulan depósitos de galactoesfingolípidos sin digerir en las glándulas ecrinas y en las células endoteliales9-11. Estos depósitos pueden provocar síntomas clínicos renales, cardíacos y cerebrovasculares, lo que conduce a una disminución de la esperanza de vida de los pacientes afectados5. El fenotipo clásico (varones con niveles indetectables o mínimos de actividad de la enzima) presenta manifestaciones graves cardíacas, renales y cerebrovasculares. Antes del desarrollo de la diálisis y el trasplante renal, la esperanza de vida era de 40 años12, pero en la actualidad se ha incrementado a una edad aproximada de 50 años13,14.
La enfermedad comienza habitualmente en la infancia o adolescencia con manifestaciones como episodios de dolor, molestias gastrointestinales (diarrea, vómitos, náuseas y dolor abdominal), proteinuria asintomática, hipohidrosis, opacidad corneal que no afecta a la visión, hipohidrosis con intolerancia al frío, al calor y al ejercicio, así como la aparición de las lesiones cutáneas conocidas como angioqueratoma corporis diffusum. Estas lesiones pasan desapercibidas en muchas ocasiones tanto para los médicos como para los pacientes o los familiares. Existen otras enfermedades de depósito lisosomal que también presentan angioqueratomas generalizados como fucosidosis, sialidosis, aspartilglucosaminuria y gangliosidosis GM1. Por ello deberá realizarse un adecuado diagnóstico diferencial. El dolor se presenta de forma paroxística, desencadenado por el ejercicio, la fatiga, el estrés emocional o los cambios bruscos de temperatura o humedad y suele ser resistente a los analgésicos convencionales. En la edad adulta, la afectación renal conduce a un estado de insuficiencia renal grave sólo tratable mediante diálisis o trasplante15,16. Las manifestaciones cardíacas incluyen hipertofia ventricular izquierda, enfermedad valvular (sobre todo insuficiencia mitral), aneurismas de la aorta ascendente, enfermedad coronaria arterial y alteraciones de la conducción. A nivel cerebrovascular, se relaciona con accidentes isquémicos transitorios, infarto cerebral, hemiparesia, vértigo y síntomas de la enfermedad vascular asociada (diplopía, disartria, nistagmo, acufenos, hemiataxia, pérdida de memoria y déficit auditivo). Entre los trastornos oculares se observa la denominada córnea verticilada, que se manifiesta hasta en el 61 % de los pacientes.
Hasta hace muy poco tiempo, el tratamiento consistía fundamentalmente en el tratamiento del dolor con analgésicos y antiepilépticos, en particular, fenitoína y carbamazepina17. Recientemente, dos compañías farmacéuticas han desarrollado una molécula de características similares que sustituye a la enzima alterada de los enfermos de Fabry: Replagal® (Transkaryotic Therapies) y Fabrazyme® (Genzyme corporation)18,19. Existe una base de datos recogida por la empresa Transkaryotic Therapies (The Fabry Outcome Survey, FOS)20 que incluye en la actualidad a más de 500 pacientes. Aproximadamente el 50 % de ellos son mujeres, aunque sólo el 43 % de ellas está en tratamiento frente al 73 % de los varones en tratamiento. Está también aumentando el número de niños incluidos en esta base de datos que hoy en día es de 74. Con ayuda de estos datos, se conoce actualmente que la progresión de la enfermedad comienza en épocas muy tempranas de la vida. Así, por debajo de los 10 años la mayoría de los niños incluidos en la FOS presentan crisis de dolor, intolerancia al calor y al frío, síntomas gastrointestinales, acufenos y vértigo. De esta forma, a los 18 años de edad alrededor del 85 % de los varones y el 65 % de las mujeres presentan síntomas. No obstante, el diagnóstico sigue siendo tardío ya que a los 10 años sólo han sido diagnosticados entre el 10 y el 20 % de los varones. Además, se empieza a constatar que las mujeres inicialmente consideradas portadoras con escasa repercusión de la enfermedad también sufren síntomas. Así, las manifestaciones cardíacas se desarrollan 10-15 años más tarde, pero no se detectan datos significativos de diferencias de incidencias acumuladas según el sexo. Esto mismo ocurre con la proteinuria que, aunque entre 10 y 15 años más tarde, también se desarrolla en las mujeres.
A nivel dermatológico, en la actualidad se tienen datos de 450 pacientes que demuestran unas diferencias significativas entre varones y mujeres. Así, la edad media de establecimiento de lesiones dermatológicas es de 15 años en varones y de 25 años en mujeres, y presentan angioqueratomas el 72 % de los varones y el 46 % de las mujeres. Además, los pacientes con lesiones dermatológicas tienen más a menudo afectación neurológica, oftalmológica, cardiovascular y renal. Actualmente, también existen resultados provisionales de biopsias cutáneas repetidas a los pacientes en tratamiento en los que se observa una reducción de los depósitos lisosomales en numerosos subtipos celulares (fibroblastos, células musculares lisas, células endoteliales, células perineurales y glándulas sudoríparas ecrinas) como signo de mejoría con el tratamiento sustitutivo.
Nuestro paciente está siendo incluido en esta base de datos. Sigue los controles y determinaciones estandarizados actualmente para los pacientes en tratamiento con niveles de globotronceramida. Aunque tradicionalmente se ha utilizado este parámetro como marcador evolutivo de la situación del paciente, hoy día está muy discutido que sea el marcador más útil para la monitorización del tratamiento debido a las fluctuaciones importantes que sufre sin relacionarse estrechamente con la respuesta terapéutica. No se ha detectado ningún efecto notable durante el mismo, excepto una pérdida de peso que contrasta con la mayoría de los pacientes en tratamiento en quienes se produce una ganancia ponderal. En el FOS se han comunicado, como efectos secundarios más importantes, la aparición de reacciones infusionales leves que aparecen hasta en el 0,7 % de infusiones. Ningún paciente ha desarrollado en la actualidad anticuerpos de tipo IgE, IgA o IgM.


