







Los tumores sebáceos son un grupo infrecuente de neoplasias. En algunos casos se han relacionado con neoplasias viscerales en pacientes con el síndrome de Muir-Torre; una variante clínica del síndrome de cáncer de colon hereditario no polipósico. El objetivo de este trabajo es revisar el diagnóstico y el seguimiento de una serie de pacientes con tumores sebáceos para comprobar cuántos de ellos cumplían criterios de síndrome de Muir-Torre.
Pacientes y métodosSe realizó una búsqueda en la base de datos del Servicio de Dermatología del Consorcio Hospital General Universitario de Valencia entre 1990 y 2005, buscando pacientes con tumores sebáceos. Se revisaron las biopsias para confirmar el diagnóstico. También buscamos datos en las historias clínicas que sugiriesen un diagnóstico de síndrome de Muir-Torre, cuando las historias estaban incompletas nos pusimos en contacto por teléfono con los pacientes.
ResultadosEncontramos 20 pacientes diagnosticados de tumores sebáceos, pero después de revisar la biopsia sólo confirmamos este diagnóstico en 12 pacientes. Dos pacientes pertenecían a una familia con antecedents de neoplasias viscerales, que cumplía los criterios clínicos de síndrome de cáncer de colon hereditario no polipósico. No hubo un seguimiento uniforme de los pacientes ni se realizaron las mismas pruebas en todos ellos.
ConclusionesEs fundamental descartar la presencia de un síndrome de Muir-Torre en pacientes con tumors sebáceos. El uso de nuevas técnicas como la detección de la inestabilidad de microsatélites o la inmunohistoquímica pueden ayudar a detectar a las familias que tienen un mayor riesgo de padecer el síndrome de Muir-Torre.
Sebaceous gland tumors are a rare type of neoplasm. In some cases they have been associated with visceral tumors in patients with Muir-Torre syndrome, a hereditary form of nonpolyposis colorectal cancer. The aim of this study was to review the diagnosis and follow-up of a series of patients with sebaceous gland tumors to assess how many met the criteria for Muir-Torre syndrome.
Patients and methodsA search was performed of records from 1990 to 2005 in the database of the Department of Dermatology of the Consorcio Hospital General Universitario de Valencia in Valencia, Spain, to identify patients with sebaceous gland tumors. The biopsy material was reviewed to confirm the diagnosis. We also searched the patient histories for information suggestive of a diagnosis of Muir-Torre syndrome; when the histories were incomplete, we contacted the patients by telephone.
ResultsWe identified 20 patients diagnosed with sebaceous gland tumors, but after reviewing the biopsy material diagnosis was only confirmed in 12. Two patients belonged to a family with a history of visceral tumors that met the clinical criteria for hereditary nonpolyposis colorectal cancer syndrome. Follow-up was not uniform in all patients and not all underwent the same tests.
ConclusionsIt is essential to rule out the presence of Muir-Torre syndrome in patients with sebaceous gland tumors. The use of new techniques such as immunohistochemistry or detection of microsatellite instability may help to identify families at increased risk of Muir-Torre syndrome.
Los tumores de diferenciación sebácea son un grupo infrecuente de lesiones con una nomenclatura compleja1. La importancia de estos tumores deriva de su asociación con neoplasias viscerales en lo que se conoce como síndrome de Muir-Torre (SMT)2,3. Este síndrome es de herencia autosómica dominante, y se caracteriza por la presencia de al menos un tumor sebáceo, junto a una neoplasia visceral maligna o bien queratoacantomas múltiples con neoplasias viscerales e historia familiar de SMT, en ausencia de otros factores predisponentes4,5. La proporción de pacientes con tumores sebáceos que desarrolla una neoplasia visceral varía según las diferentes series entre un 13,96 y un 42%7.
En los años 80 aparecieron casos de SMT en familias diagnosticadas de síndrome de cáncer de colon hereditario no polipósico o síndrome de Lynch8. Posteriormente se comprobó que el SMT y el síndrome de Lynch compartían el mismo defecto genético9,10 en unas proteínas que se encargan de reparar los desapareamientos de nucleótidos en el ADN, por eso actualmente se considera el SMT como una forma clínica del síndrome de Lynch11. La sospecha de síndrome de Lynch se establece a partir de unos criterios clínicos que evalúan los antecedentes personales y familiares de neoplasias, y que se conocen como criterios de Amsterdam12,13 y de Bethesda14.
En estos pacientes los tumores viscerales se localizan, sobre todo, en el colon, pero también en el endometrio, el ovario, el estómago, el intestino delgado, el uréter, la pelvis renal y el cerebro12. Otras características del SMT son la presencia de tumores múltiples, su presentación a una edad más temprana y, en algunos casos, un mejor pronóstico que cuando esos mismos tumores aparecen de forma espontánea11.
El objetivo de este trabajo es revisar el diagnóstico y el seguimiento de una serie de pacientes con tumores sebáceos para comprobar cuántos de ellos cumplían criterios de SMT.
Pacientes y métodosSe realizó una búsqueda en la base de datos de dermatopatología del Servicio de Dermatología del Hospital General Universitario de Valencia entre los años 1990 y 2005, utilizando los términos adenoma sebáceo, epitelioma sebáceo, carcinoma basocelular con diferenciación sebácea, se- baceoma y carcinoma sebáceo. Excluimos de la búsqueda otras neoplasias sebáceas no relacionadas con el SMT como la hiperplasia sebácea, el nevus sebáceo de Jadassohn o las neoplasias mixtas de diferenciación sebácea y apocrina. Las biopsias se revisaron para confirmar el diagnóstico, buscando signos de diferenciación sebácea15 (tabla 1) y seleccionando aquellas biopsias con datos inequívocos de tumor sebáceo.
Criterios histopatológicos de diferenciación sebácea
| 1. Células con morfología de sebocitos similares a los de la glándula sebácea normal |
| 2.Conductos que queratinizan de manera similar a como lo hace el conducto sebáceo |
| 3.Sebo |
| 4.Sebolema o vaina del sebo |
fuente: Requena C, et al15.
Revisamos las historias clínicas de los pacientes seleccionados, buscando datos sobre la historia familiar y personal de cáncer que fueran compatibles con los criterios de Amsterdam o de Bethesda (tabla 2). También buscamos datos sobre las exploraciones complementarias realizadas en cada caso y la evolución clínica. En los casos en los que no había datos en la historia clínica llamamos al paciente por teléfono.
Criterios diagnósticos en el síndrome de Lynch
| Criterios de Amsterdam I |
| Debe haber al menos tres parientes con cáncer colorrectal |
| Un familiar debe ser pariente en primer grado de los otros dos |
| Al menos dos generaciones deben estar afectadas |
| Al menos un tumor debe ser diagnosticado antes de los 50 años |
| Deberá excluirse la poliposis adenomatosa familiar |
| Criterios de Amsterdam II |
| Igual que los criterios de Amsterdam I, pero se incluyen pacientes con otros tumores asociados al SCCHNP (endometrio, ovario, intestino delgado, uréter o pelvis renal) |
| Criterios de Bethesda |
| Cáncer colorrectal en un paciente menor de 50 años |
| Presencia de tumores colorrectales u otros tumores asociados al SCCHNP*, sincrónicos o metacrónicos independientemente de la edad |
| Cáncer colorrectal con una histología de células en anillo de sello o mucinosa, linfocitos infiltrando el tumor, reacción linfocitaria peritumoral Crohn-like o patrón de crecimiento medular en pacientes menores de 60 años |
| Pacientes con cáncer colorrectal, y un pariente de primer grado que presenta un tumor asociado al SCCHNP*, con uno de los tumores diagnosticados antes de los 50 años |
| Pacientes con cáncer colorrectal con dos o más parientes de primer o segundo grado con un tumor asociado al SCCHNP, independientemente de la edad. |
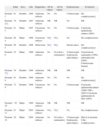
Entre 1990 y 2005 encontramos 20 pacientes con tumores sebáceos; sin embargo, al revisar la histología, sólo 12 pacientes tenían criterios histológicos claros de tumor sebáceo. En los otros 8 pacientes las biopsias correspondían a otro tipo de tumores no relacionados con el SMT, los datos correspondientes a estos pacientes aparecen en la tabla 3. En dos casos (pacientes d y e), las biopsias eran parciales, y cuando se extirparon por completo el diagnóstico coincide con el nuestro. En este grupo de pacientes también revisamos las historias clínicas y encontramos que cuatro de ellos (pacientes a, b, c y g) habían sido sometidos a exploraciones complementarias para descartar la presencia de neoplasias.
Pacientes con diagnóstico de tumor sebáceo no confirmado en la revisión de la biopsia
| Edad | Sexo | Año | Diagnóstico inicial | Diagnóstico revisado | Exploraciones | |
| Paciente a | 67 | Hombre | 1990 | Epitelioma sebáceo | Espiroadenoma | Enema opaco |
| Paciente b | 64 | Hombre | 1991 | Adenoma sebáceo | Hiperplasia sebácea | Colonoscopia |
| Paciente c | 79 | Hombre | 1992 | Carcinoma sebáceo | Carcinoma epidermoide | Colonoscopia |
| Paciente d | 59 | Hombre | 1992 | Epitelioma sebáceo | Carcinoma epidermoide | No |
| Paciente e | 85 | Mujer | 1993 | Carcinoma espinocelular frente a carcinoma sebáceo | Enfermedad de Bowen | No |
| Paciente f | 86 | Mujer | 1995 | Carcinoma sebáceo | Hidroadenocarcinoma apocrino | No |
| Paciente g | 65 | Hombre | 1996 | Carcinoma basocelular con diferenciación sebácea | Carcinoma basocelular de células claras | Enema opaco |
| Paciente h | 95 | Mujer | 1997 | Adenoma sebáceo | Hiperplasia sebácea | No |
Los datos de los pacientes con tumores sebáceos confirmados aparecen recogidos en la tabla 4. Se trata de 6 hombres y 6 mujeres con una edad media de 68 años. La mayoría de los tumores sebáceos corresponden a adenomas sebáceos y sólo un caso (paciente 6) tiene tumores múltiples. En dos casos (pacientes 7 y 10), los pacientes fueron vistos en un ambulatorio externo y no fueron remitidos posteriormente al hospital, por lo que no sabemos cuál fue su evolución, ni las exploraciones complementarias que se realizaron. En otro caso (paciente 2), no había ningún teléfono de contacto, por lo que tampoco pudimos saber la evolución, ni si existían antecedentes familiares o personales de cáncer, ya que éstos no se habían recogido en la historia. Otros tres pacientes (pacientes 4, 5 y 11), no tenían datos en la historia clínica sobre los antecedentes personales o familiares de cáncer; sin embargo, en estos tres casos pudimos localizar a los pacientes por teléfono y comprobamos que no cumplían los criterios diagnósticos de síndrome de Lynch.
Pacientes con tumores sebáceos confirmados en la revisión de la biopsia
| Edad | Sexo | Año | Diagnóstico | AP de cáncer | AF de cáncer | Exploraciones | Evolución | |
| Paciente 1 | 61 | Hombre | 1994 | Adenoma sebáceo | No | No | Colonoscopia | Sin complicaciones* |
| Paciente 2** | 70 | Hombre | 1997 | Adenoma sebáceo | NR | NR | No | NR |
| Paciente 3 | 81 | Mujer | 1997 | Carcinoma sebáceo | No | No | Colonoscopia | Carcinoma epidermoide cutáneo (2003) |
| Paciente 4 | 86 | Mujer | 1998 | Carcinoma sebáceo | No* | No* | No | Sin complicaciones |
| Paciente 5 | 66 | Hombre | 1998 | Sebaceoma | No* | No* | Enema opaco | Sin complicaciones* |
| Paciente 6 | 52 | Mujer | 1998 | Adenoma sebáceo | No | Sí (colon y endometrio) | Colonoscopia Exploración ginecológica | Carcinoma de endometrio (1999), sebomatricoma (2002) Colonoscopia bianual |
| Paciente 7** | 76 | Hombre | 1998 | Adenoma sebáceo | NR | NR | NR | NR |
| Paciente 8 | 61 | Hombre | 1998 | Adenoma sebáceo | No | No | No | Sin complicaciones* |
| Paciente 9 | 72 | Hombre | 1998 | Carcinoma sebáceo | No | No | Colonoscopia | Carcinoma epidermoidecutáneo (1999) VIH+(2001) Carcinoma epidermoide de labio (2003) |
| Paciente 10 | "93 | Mujer | 1999 | Adenoma sebáceo | NR | NR | NR | NR |
| Paciente 11 | 81 | Mujer | 2002 | Adenoma sebáceo | No* | No* | No | Sin complicaciones |
| Paciente 12 | 28 | Mujer | 2004 | Adenoma sebáceo | No | Sí (colon y endometrio) | Colonoscopia Exploración ginecológica | Hija de la paciente 6 Sin complicaciones |
Año: año de diagnóstico; AF: antecedentes familiares; AP: antecedentes personales; NR: no registrado.
Se realizaron exploraciones complementarias para descartar neoplasias en 6 de los 12 pacientes (pacientes 1, 3, 5, 6, 9 y 12). En dos casos (pacientes 4 y 8), los pacientes rechazaron la realización de exploraciones complementarias.
En el resto de los pacientes no se especifica la razón por la que no se realizan las exploraciones (pacientes 2 y 11), o bien no hay datos sobre los pacientes (pacientes 7 y 10).
Sólo había antecedentes familiares de cáncer en dos pacientes (pacientes 6 y 12) que pertenecían a una misma familia, con una historia familiar que además cumplía los criterios de Amsterdam y que exponemos a continuación con más detalle.
Pacientes 6 y 12La paciente 6 acudió a la consulta, en septiembre de 1998, por una pequeña pápula erosionada de color amarillento en la mejilla derecha, que fue diagnosticada clínicamente de carcinoma basocelular (fig. 1). En el análisis histopatológico de la lesión, se observó una neoplasia localizada en la dermis superficial, formada en su mayoría por sebocitos maduros que se agrupan de una forma desorganizada (fig. 2), que se diagnosticó de adenoma sebáceo. La historia familiar tenía varios casos de cáncer de colon y endometrio en familiares de primer y segundo grado (fig. 3) y cumplía los criterios de Amsterdam. La paciente se sometió a un programa de cribado para el cáncer de colon, con colonoscopias y exploraciones ginecológicas bianuales. Durante la realización de la primera exploración ginecológica, en enero de 1999, se detectó una neoplasia de endometrio que fue intervenida ese año mediante una histerectomía con doble anexectomía. Sus familiares de primer grado se sometieron también a un programa de cribado, aunque uno de sus hermanos lo rechazó. Durante el seguimiento de la familia, su hija (paciente 12; paciente III: 2) (fig. 3) presentó una lesión en la nariz clínicamente parecida a la de su madre, pero más pequeña, que fue extirpada en 2004 con 28 años de edad. La biopsia de la lesión mostró unos nódulos mal delimitados localizados en la dermis media, formados en su mayoría por sebocitos maduros y sin conexión con un infundíbulo en los cortes seriados (fig. 4), que también fueron diagnosticados como adenoma sebáceo. Ese año la familia fue remitida a un centro de referencia para realizar un estudio genético, detectándose una deleción en los exones 1 y 2 del gen MSH2 en la paciente 6 y en sus dos hijos, aunque uno de ellos no había presentado tumores en el momento de hacerse las pruebas (paciente III:1) (fig. 3). Dos de sus hermanos rechazaron hacerse las pruebas. No tenemos información sobre el resto de sus familiares, porque el seguimiento se realizaba en otros centros.

.")
 y 12.")
.")
El primer paso en el diagnóstico del SMT es el hallazgo histopatológico de un tumor sebáceo relacionado con este síndrome. Dentro de este grupo de tumores no se incluyen tumores como la hiperplasia sebácea, el nevus sebáceo y otras neoplasias que derivan de la unidad folículo-sebáceo-apocrina, que pueden presentar diferenciación sebácea de forma focal. El diagnóstico diferencial también incluye otras lesiones que presenten células claras, como el carcinoma basocelular de células claras, el carcinoma espinocelular de células claras, la enfermedad de Paget, la enfermedad de Bowen pagetoide, el hidroadenocarcinoma de células claras, el carcinoma siringoide de células claras o las metástasis cutáneas de un carcinoma renal. Ese diagnóstico diferencial puede ser complicado, de hecho en nuestra serie de pacientes una proporción importante (40 %) corresponde a otros tumores de células claras o a hiperplasias sebáceas.
Una de las causas que puede haber influido en la mala interpretación de estas biopsias es la utilización de conceptos mal definidos como el de carcinoma basocelular de diferenciación sebácea. Para algunos autores16, la mayoría de los casos descritos corresponden a carcinomas basocelulares de células claras, como en nuestra serie (paciente g), o a glándulas sebáceas atrapadas por el crecimiento del tumor. Una forma de evitar confusiones sería usar conceptos que unifiquen los diferentes nombres usados en los tumores sebáceos en entidades bien definidas, como el concepto de sebomatricoma planteado por Sánchez Yus et al17, y que englobaría a todos los tumores benignos con diferenciación sebácea. Otros autores, como Simón et al18, también apoyan el uso de este término.
Otro factor que puede influir en el diagnóstico erróneo es que la biopsia sea pequeña o esté artefactada, como en dos de nuestros casos (pacientes d y e), en los que al extirpar el tumor de forma completa se llegó al diagnóstico correcto.
Antes de hacer un diagnóstico histológico de tumor sebáceo hay que tener en cuenta que esto puede implicar la realización de exploraciones complementarias para descartar neoplasias internas, como fue el caso de cuatro de nuestros pacientes (pacientes a, b, c y g), con el consiguiente gasto innecesario y el estrés emocional que para los pacientes conlleva la posibilidad de tener una neoplasia oculta.
Después de la histología, el siguiente paso es la realización de una historia clínica exhaustiva buscando antecedentes familiares y personales de cáncer6,19 que sean compatibles con los criterios de Amsterdam o de Bethesda. En nuestra serie de pacientes con tumores sebáceos, sólo el 50 % de los casos tenía registrado en la historia clínica los antecedentes familiares o personales de cáncer. Esto puede deberse a que las historias estaban mal cumplimentadas o a que se desconocían las implicaciones clínicas que tenía el diagnóstico histopatológico. Por eso, además de las historias clínicas, nos pusimos en contacto por teléfono con los pacientes para comprobar sus antecedentes.
-Encontramos que sólo dos de nueve pacientes (pacientes 6 y 12, 22,2 %), que pertenecían a una misma familia, tenían unos antecedentes sugestivos de síndrome de Lynch. Aunque nuestra serie es muy pequeña, esta proporción es similar a la comunicada en un artículo reciente de Ponti et al6, en la que encuentran criterios de SMT en 5 de 36 pacientes con neoplasias sebáceas (13,9 %) y es significativamente menor que la de otra serie publicada por Finan y Conolly7 en los años 80, en la que encuentran estos criterios en 25 de 59 pacientes (42 %). Esta diferencia se puede deber a que Finan y Conolly incluyen en su serie pacientes con entidades confusas, como el carcinoma basocelular de diferenciación sebácea (2 pacientes), o pacientes que presentan lesiones«adenomatosas»(sic) con características de hiperplasia sebácea, epitelioma o perlas córneas (9 pacientes), por lo que es probable que alguno de esos pacientes no tuviese un tumor sebáceo relacionado con el SMT, y los resultados que encontraron no sean válidos. No obstante, también hay que tener en cuenta las limitaciones de nuestro estudio, ya que la muestra es pequeña y pertenece a un único centro, lo que impide tener datos suficientemente precisos de la incidencia. Además, al ser un estudio retrospectivo y no tener todos los pacientes un seguimiento homogéneo, la incidencia de los tumores puede estar subestimada.
El árbol genealógico de la familia de nuestro estudio (fig. 3) cumple los criterios de Amsterdam II y es un claro ejemplo de enfermedad autosómica dominante con expresión variable y penetrancia incompleta. La expresión variable se refiere a la diferente expresión que tiene un mismo defecto genético en diferentes individuos; por ejemplo, sólo nuestras dos pacientes tienen tumores sebáceos, mientras que el resto de los familiares enfermos presenta otro tipo de neoplasias. La penetrancia incompleta se refiere a que no todos los individuos que heredan el gen van a desarrollar la enfermedad, el mejor ejemplo de esta característica es uno de los miembros de la familia (II:8). Mientras que uno de sus hijos (III:8) y su padre (I:3) han tenido cáncer de colon, él no presentaba la enfermedad. Esto se debe a que estos individuos son heterocigotos para la mutación, y conservan una copia sana del gen afecto, factores ambientales hacen que esa copia no se exprese y no se desarrolle la neoplasia.
En ocasiones los antecedentes familiares no pueden ser valorados, como por ejemplo en caso de adopción, falsa paternidad, desconocimiento de las causas de muerte en la familia, por la penetrancia incompleta en familias pequeñas o en el caso de mutaciones de novo20, por lo que la ausencia de antecedentes familiares no descarta del todo la presencia de una mutación en las proteínas reparadoras del ADN.
Para descartar esa mutación los protocolos más recientes recomiendan determinar la inestabilidad de microsatélites en los tumores del paciente o de sus familiares6,19. Los microsatélites son secuencias repetitivas de dinucleótidos, trinucleótidos o tetranucleótidos distribuidos en el genoma, que se alteran de forma significativa cuando los genes encargados de reparar los desapareamientos no funcionan.
Sin embargo, puede ser más práctico el uso de la inmunohistoquímica para detectar la proteína deficiente, ya que se ha demostrado una buena correlación entre la inestabilidad de microsatélites y la inmunohistoquímica21. Las dos proteínas que se alteran con mayor frecuencia son la MLH1 y, sobre todo, la MSH222, como en nuestros casos, aunque también se han detectado casos asociados al gen MSH623. Existen reactivos para detectar estas tres proteínas mediante la inmunohistoquímica, en los pacientes afectos se observa una falta de tinción de la proteína deficiente. Si estas pruebas de cribaje muestran alteraciones se busca la mutación secuenciando el gen y, si se encuentra, entonces se examina a los familiares del paciente para detectar a los portadores y someterlos a un programa de cribado de cáncer. El hecho de no encontrar una mutación no excluye totalmente a la familia, ya que puede tener un defecto genético que no se ha detectado hasta el momento11, y si la historia clínica es sugestiva, el paciente y sus familiares de primer grado deberían someterse a programas de cribado de cáncer.
Estas técnicas no estaban disponibles cuando se diagnosticaron nuestros pacientes, por lo que para descartar la presencia de neoplasias asociadas se realizaron colonoscopias o enemas opacos en la mitad de nuestros casos, en la mayoría de ellos de forma puntual. Teniendo en cuenta que hasta en un 22 % de los casos de SMT la neoplasia interna asociada aparece después de la detección del tumor sebáceo24, es probable que alguno de nuestros casos pudiese desarrollar un tumor años después de la realización puntual de la colonoscopia o el enema opaco, ya que el seguimiento fue irregular y en algún caso el estudio fue incompleto. No obstante, debido a la elevada edad media de nuestros pacientes (68 años), esa probabilidad parece ser baja. En nuestra serie sólo sometimos a un programa de cribado de cáncer de forma regular a los dos pacientes (pacientes 6 y 12) con antecedentes sugestivos de síndrome de Lynch, que además eran los más jóvenes, y aparte de sus familiares de primer grado, el resto de los familiares estaban controlados en otros centros.
Los programas de cribado, en el caso del síndrome de Lynch, incluyen la realización de colonoscopias cada uno o dos años, empezando a partir de los 20–25 años, y en el caso de las mujeres exploraciones ginecológicas con ecografía transvaginal, biopsia endometrial por aspirado y determinaciones de CA-125 en suero a partir de los 30 años, también de forma anual. La realización de otras exploraciones depende de la historia familiar de cáncer24,25.
En el caso del SMT, algunos autores han propuesto la realización de mamografías cada uno o dos años, empezando en el momento del diagnóstico, hasta los 50 años, y a partir de entonces cada año19,26; sin embargo, hay que tener en cuenta que el cáncer de mama, como otros cánceres descritos en el SMT4,24, no se ha podido relacionar con el síndrome de Lynch25, por lo que su presentación en un paciente aislado puede ser incidental. También se ha propuesto la realización de una tomografía axial computarizada (TAC) de abdomen y pelvis cada 2–5 años19, porque el 35 % de los casos de tumores abdominales en el SMT corresponde a tumores distintos al cáncer de colon27.
El papel del dermatólogo es fundamental en el diagnóstico de estos pacientes. Los datos de nuestra serie sugieren que en algunos casos se subestiman estas lesiones y no se tienen en cuenta los antecedentes familiares de cáncer ni se realizan exploraciones complementarias para descartar tumores.
La utilización de nuevas técnicas como la inestabilidad de microsatélites, la inmunohistoquímica y la secuenciación de los genes afectados permite establecer mejor el riesgo de estos pacientes de padecer un cáncer, pero no sustituye a los datos que se obtienen de la historia clínica.
Conflicto de interesesDeclaramos no tener ningún conflicto de intereses.
Los autores declaran no tener ninguna fuente de financiación.


