


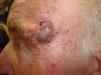

Un varón de 82 años, hipertenso, con hernia de hiato y osteoporosis, acudió a la consulta por una lesión localizada en la sien izquierda, de 4 meses de evolución, dolorosa a la presión, no sangrante ni ulcerada.
Exploración físicaSe objetivó un tumor excrecente de 2cm de diámetro, de bordes bien delimitados y coloración eritemato-violácea, situado en la sien izquierda, que alcanzaba en su borde inferior la cola de la ceja (fig. 1). Al tacto se encontraba adherido a planos profundos y no se palpaban adenopatías locorregionales.
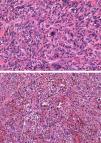
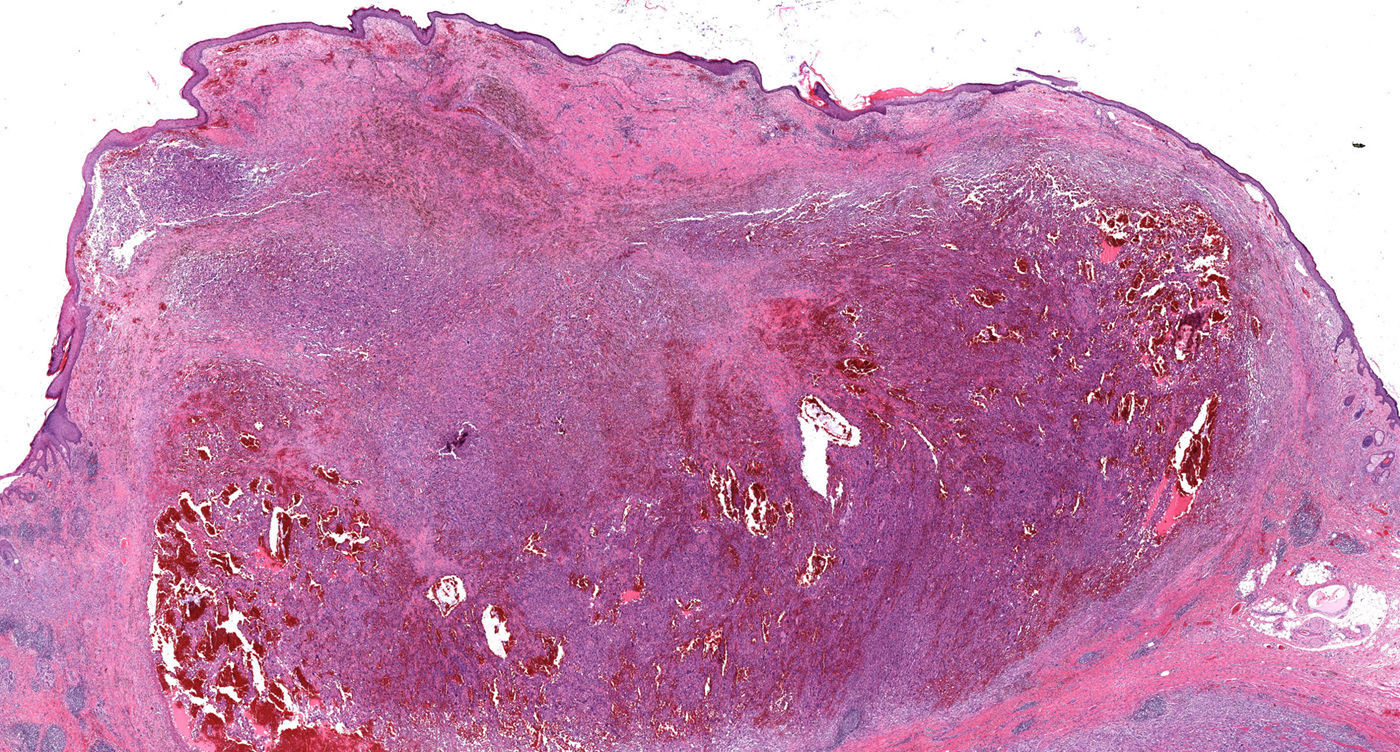
Histopatología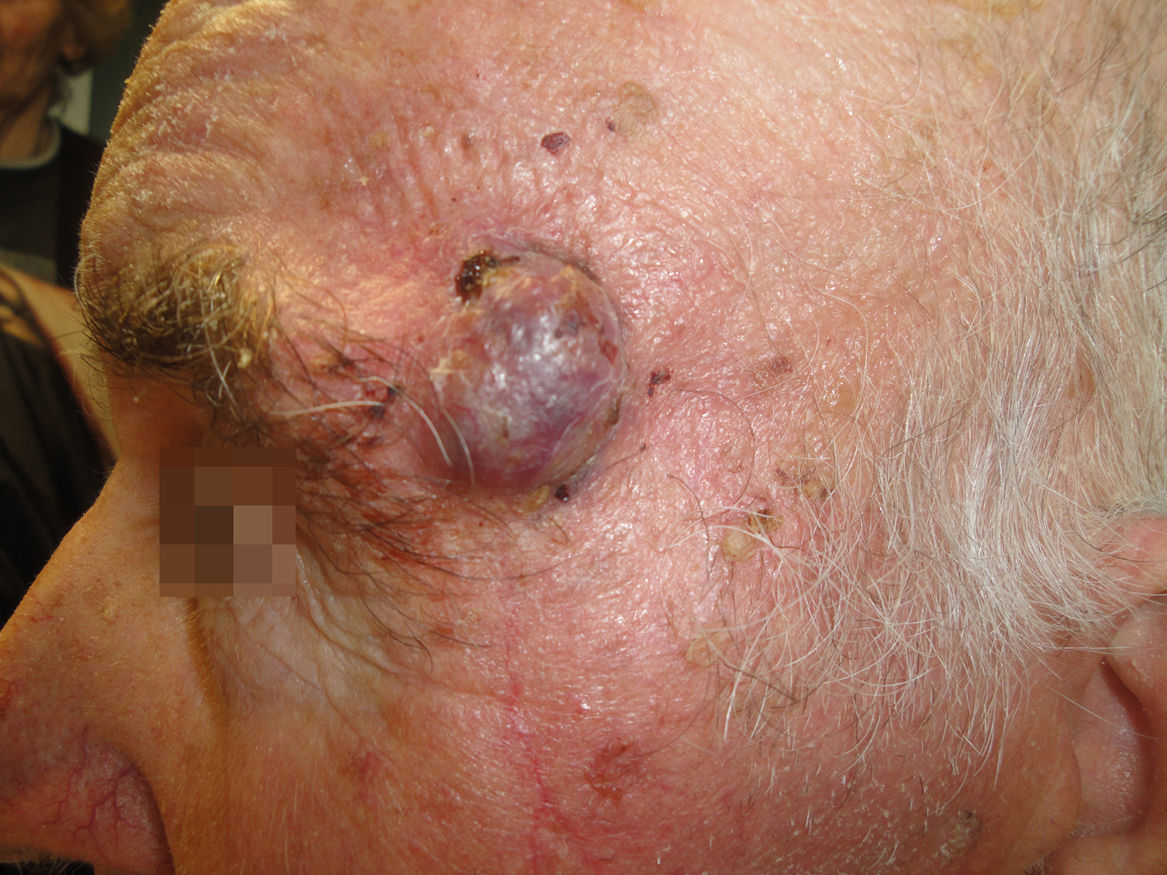
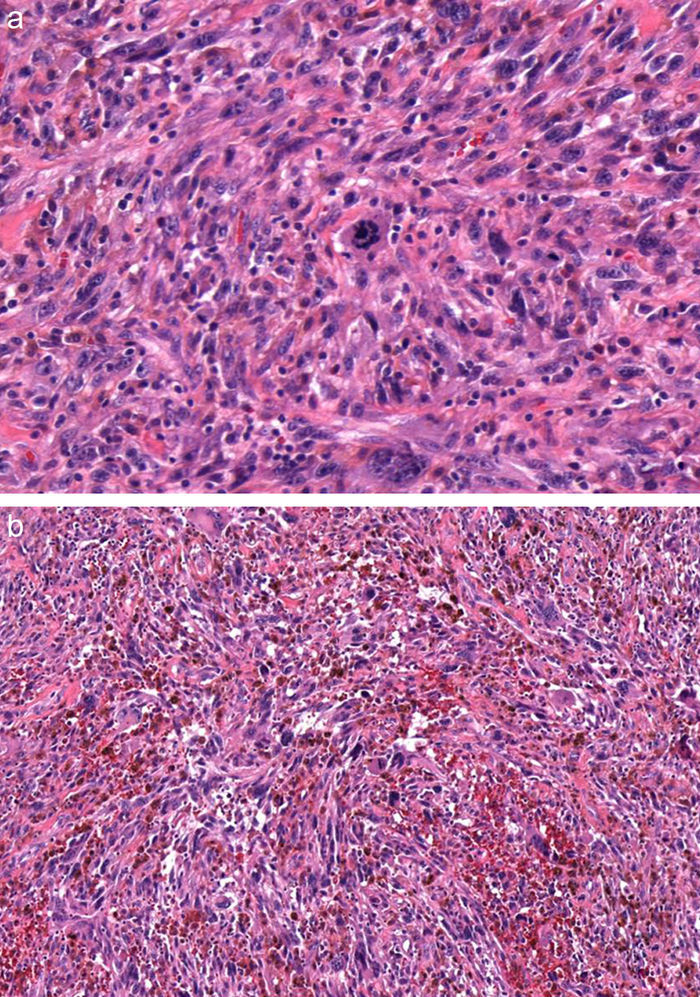
En la pieza de la biopsia escisional, la tinción con hematoxilina-eosina reveló una epidermis ortoqueratósica, sobre un infiltrado dérmico de células pleomórficas elongadas, entremezcladas con otras de núcleos redondeados e irregulares, y abundantes mitosis aberrantes (fig. 2 y fig. 3a). Este infiltrado presentaba una disposición estoriforme sobre un estroma fibroso colagenizado, con llamativas estructuras vasculares (fig. 3b). Las tinciones inmunohistoquímicas demostraron positividad para CD68 y CD31, positividad focal para S100, positividad aislada para vimentina y un índice de proliferación Ki67, cercano al 70%. El resto de tinciones, tales como actina de músculo liso, desmina, citoqueratinas AE1/AE3, CD 34, CD10, antígeno leucocitario común, HMB45 y Melan A resultaron negativas.
¿Cuál es su diagnóstico?
DiagnósticoSarcoma dérmico pleomórfico.
Evolución y tratamientoLa lesión fue extirpada sin obtener márgenes quirúrgicos libres, por lo que se realizó cirugía de Mohs diferida, con exéresis total tras un único pase. El defecto fue cubierto mediante un injerto de piel total.
El estudio de extensión demostró una adenopatía retroparotídea izquierda, que resultó patológica en la punción practicada. El caso fue evaluado en el comité de tumores, descartándose tratamientos complementarios. Tras 18 meses de seguimiento, el paciente no ha presentado recidiva local ni modificaciones en el curso de la enfermedad.
ComentarioLos sarcomas suponen las neoplasias de origen mesenquimal más frecuentes en el adulto. Su clasificación y pronóstico están determinados por su estirpe celular.
Los recientes avances en inmunohistoquímica y genética molecular han supuesto una disminución progresiva en la incidencia del antes conocido como histiocitoma fibroso maligno, en favor de otras entidades más específicas1,2. En la última clasificación de tumores óseos y de partes blandas de la World Health Organization (WHO) en 2013, se sustituye dicho término por el de sarcoma pleomórfico indiferenciado1,3. Este es un diagnóstico de exclusión para neoplasias fibrohistiocitarias sin una línea de diferenciación evidente mediante las técnicas diagnósticas disponibles actualmente. Debemos evitar la confusión del ahora sarcoma pleomórfico indiferenciado, con el sarcoma dérmico pleomórfico de nomenclatura similar4–6.
El sarcoma dérmico pleomórfico suele manifestarse en varones en la séptima u octava década de la vida, como un nódulo dérmico de crecimiento rápido, con zonas necróticas y hemorrágicas, preferentemente en regiones fotoexpuestas como la cabeza y el cuello4,5. Característicamente, presenta células epitelioides pleomórficas y células fusiformes atípicas con numerosas mitosis, y extensas áreas de ulceración y necrosis, así como extensión hacia planos profundos e invasión vascular y perineural, lo que lo distingue del fibroxantoma atípico4–6. La inmunohistoquímica resulta clave para el diagnóstico al excluir otras entidades como tumores de estirpe melanocítica, tumores malignos de vaina nerviosa, carcinomas escamosos poco diferenciados, angiosarcomas, leiomiosarcomas o el carcinoma de células de Merkel2,6.
La diseminación metastásica se estima en un 10%, y su recidiva local en un 30%4–6. No se dispone de datos de series amplias debido a los múltiples cambios de nomenclatura.
El tratamiento de elección es la cirugía convencional con márgenes o la cirugía de Mohs4,6. Al igual que en otros tumores fibrohistiocitarios no existen recomendaciones consenso acerca del tratamiento de la enfermedad metastásica o su seguimiento.
Con este caso incidimos en la necesidad de conocer y aplicar la nueva clasificación publicada por la WHO, que nos permitirá homogeneizar el diagnóstico de las neoplasias de partes blandas, y con ello mejorar el manejo clínico del paciente. Además, resaltamos la importancia de la inmunohistoquímica en el diagnóstico de estos tumores, y la necesidad de realizar un estudio de extensión, ante la posibilidad de metástasis a distancia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Al Dr. Rodríguez Peralto y la Dra. Enguita Valls del Servicio de Anatomía Patológica del Hospital Universitario 12 de Octubre, por su inestimable ayuda.


