




HISTORIA CLÍNICA
Un varón de 40 años, con antecedentes personales y familiares (madre y dos hermanos) de enfermedad de von Recklinghausen consultó por la presencia de un tumor sacrococcígeo doloroso de 5 meses de evolución, con un rápido crecimiento y sin sintomatología neurológica motora ni sensitiva acompañante.
EXPLORACIÓN FÍSICA
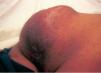
El paciente tenía deformidad de la hemicara izquierda, con irritación conjuntival y lagoftalmo ipsolateral, secundarios a un neurofibroma que había requerido varias intervenciones entre los 2 y los 18 años. Además tenía múltiples manchas «café con leche» y neurofibromas en el tórax, los miembros superiores e inferiores y la cara. En la zona sacrococcígea se observó un gran tumor de 10 × 11 cm de diámetro, eritematoso, caliente, de consistencia dura elástica con zonas fluctuantes, doloroso al tacto y espontáneamente (fig. 1). En el tacto rectal se detectó que la ampolla rectal estaba libre y que la pared ventral del recto estaba abombada por el tumor.
EXPLORACIONES COMPLEMENTARIAS
El examen oftalmológico con lámpara de hendidura mostró un nódulo de Lisch en el iris del ojo derecho. El hemograma, las radiografías torácicas y lumbares y el electrocardiograma fueron normales.
La resonancia magnética (RM) nuclear lumbosacra con gadolinio puso de manifiesto una gran masa, de unos 10 cm de diámetro máximo, que asentaba por debajo de las últimas vértebras sacras (fig. 2). Por delante, contactaba con el recto. Tenía una señal heterogénea en su interior, con áreas sólidas y quísticas. En el sector posterior e inferior del tejido celular subcutáneo perineal, junto a la escotadura ciática izquierda, se observaba una lesión de unos 30 mm con características similares. Se realizó exéresis y estudio histológico del tumor (fig. 3).
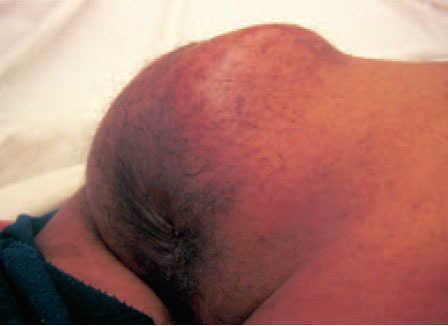
Fig. 1.—Gran tumor sacrococcígeo doloroso de rápido crecimiento.
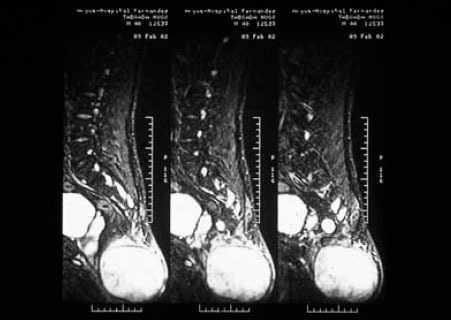
Fig. 2.—RM de la zona lumbosacra que muestra la extensión del tumor.
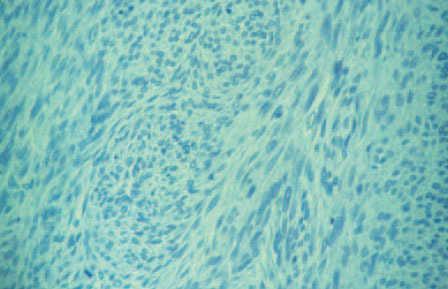
Fig. 3.—Detalle histológico del tumor. (Hematoxilina-eosina, ×10.)
DIAGNÓSTICO
Tumor maligno de la vaina nerviosa del nervio periférico (schwannoma maligno).
HISTOPATOLOGÍA
En el examen histológico se observó una neoplasia maligna constituida por células fusiformes de aspecto neuroide dispuestas en haces entrelazados y ondulados ocupando la dermis. Existían amplias áreas de necrosis, hemorragias, alto índice mitótico con mitosis atípicas y degeneración mixoide. La neoplasia, con bordes parcialmente lobulados, infiltraba hasta el tejido celular subcutáneo, sin que pudiera precisarse el límite de la lesión. La piel perianal no estaba comprometida.
Con tinciones para reticulina se observaron fibrillas rodeando grupos celulares y perivasculares.
EVOLUCIÓN Y TRATAMIENTO
Se extirpó un gran tumor, encapsulado, de más o menos 10-12 cm de diámetro mayor. El extremo superior estaba por debajo del coxis y el inferior contactaba con el músculo elevador del ano y el esfínter anal; anteriormente contactaba con el recto, sin infiltrarlo. Se observaron cuatro raíces nerviosas del plexo sacro con filetes nerviosos derechos e izquierdos que formaban parte del tumor. El tratamiento incluyó antibioterapia y analgésicos posquirúrgicos. Meses después el paciente sufrió una recidiva tumoral y metástasis pulmonares, y falleció al poco tiempo.
COMENTARIO
La alteración genética subyacente en la neurofibromatosis tipo 1 (NF-1) radica en el gen de la neurofibromina (localizado en el brazo largo del cromosoma 17q11.2), que actúa como un gen supresor tumoral1. La pérdida de heterozigosidad en las células tumorales de los neurofibromas de estos individuos favorece su malignización, con aparición de neurofibrosarcomas en el 4,5 % de pacientes con NF-1. El más frecuente es el schwannoma maligno o tumor maligno de la vaina nerviosa, cuya incidencia en la población general es 0,001 %. El tumor maligno de la vaina de los nervios periféricos es una neoplasia poco común, que comprende aproximadamente el 10 % de todos los sarcomas de tejidos blandos. Ha recibido diferentes nombres, como schwannoma maligno, neurofibrosarcoma, sarcoma neurogénico y tumor maligno de la vaina de los nervios periféricos, siendo este último el más aceptado, puesto que esta neoplasia puede tener características de cualquiera de los tipos celulares que componen la vaina de los nervios periféricos: células de Schwann, células perineurales o fibroblastos2-7.
En cuanto a la presentación clínica, el tumor puede ser superficial o profundo; los de localización profunda se ubican excéntricamente en nervios proximales o en las raíces nerviosas raquídeas comunicantes sensitivas. La lesión suele crecer con rapidez y tornarse dolorosa8; se manifiesta como un tumor de tamaño variable que puede asociar o no signos y síntomas neurológicos. El patrón histológico también es variable: mesenquimatoso, glandular y epitelial, similar a un neuroepitelioma9. En nuestro paciente el patrón histológico fue mesenquimatoso.
El tumor tiende a recurrir y a ocasionar metástasis pulmonares (en el 39 % de los casos) y en el tejido blando en un tiempo medio de 20 meses. El porcentaje de recaídas y metástasis a los 5 años es del 49 %; la supervivencia global a los 5 años oscila entre el 34 y el 53 %.
Los factores que afectan adversamente el pronóstico son la edad temprana de aparición, el mayor tamaño tumoral (> 10 cm) y la extensión de la resección10. El tratamiento de elección es la resección quirúrgica. Los resultados con quimioterapia y radioterapia son desalentadores.


