





INTRODUCCION
El tricoepitelioma es una neoplasia cutánea benigna con diferenciación hacia células germinativas foliculares. Se distinguen tres formas clínicas de tricoepitelioma: solitario, múltiple, que en la gran mayoría de los casos es familiar con herencia autosómica dominante y desmoplásico. Histopatológicamente, las lesiones solitarias y múltiples de tricoepitelioma son indistinguibles entre sí, mientras que el tricoepitelioma desmoplásico (TD), siendo también un tricoblastoma, presenta características clínicas e histológicas peculiares.
DESCRIPCION DE LOS CASOS
Caso 1
Una mujer de 27 años de edad, sin antecedentes personales ni familiares de interés, consultó por una lesión cutánea asintomática de aproximadamente 3 años de evolución, que había aumentado de tamaño de forma lenta y progresiva. Durante los primeros meses recibió tratamiento con preparados de ácido salicílico sin obtener ninguna respuesta clínica, lo que motivó la suspensión de dicho tratamiento.
A la exploración física se apreciaba una lesión única, anular de 7 mm de diámetro, localizada en la mejilla derecha (fig. 1), con bordes elevados y centro ligeramente deprimido, de consistencia aumentada y no adherida a planos profundos. No se palparon adenopatías regionales y el resto de la exploración cutánea fue compatible con la normalidad.
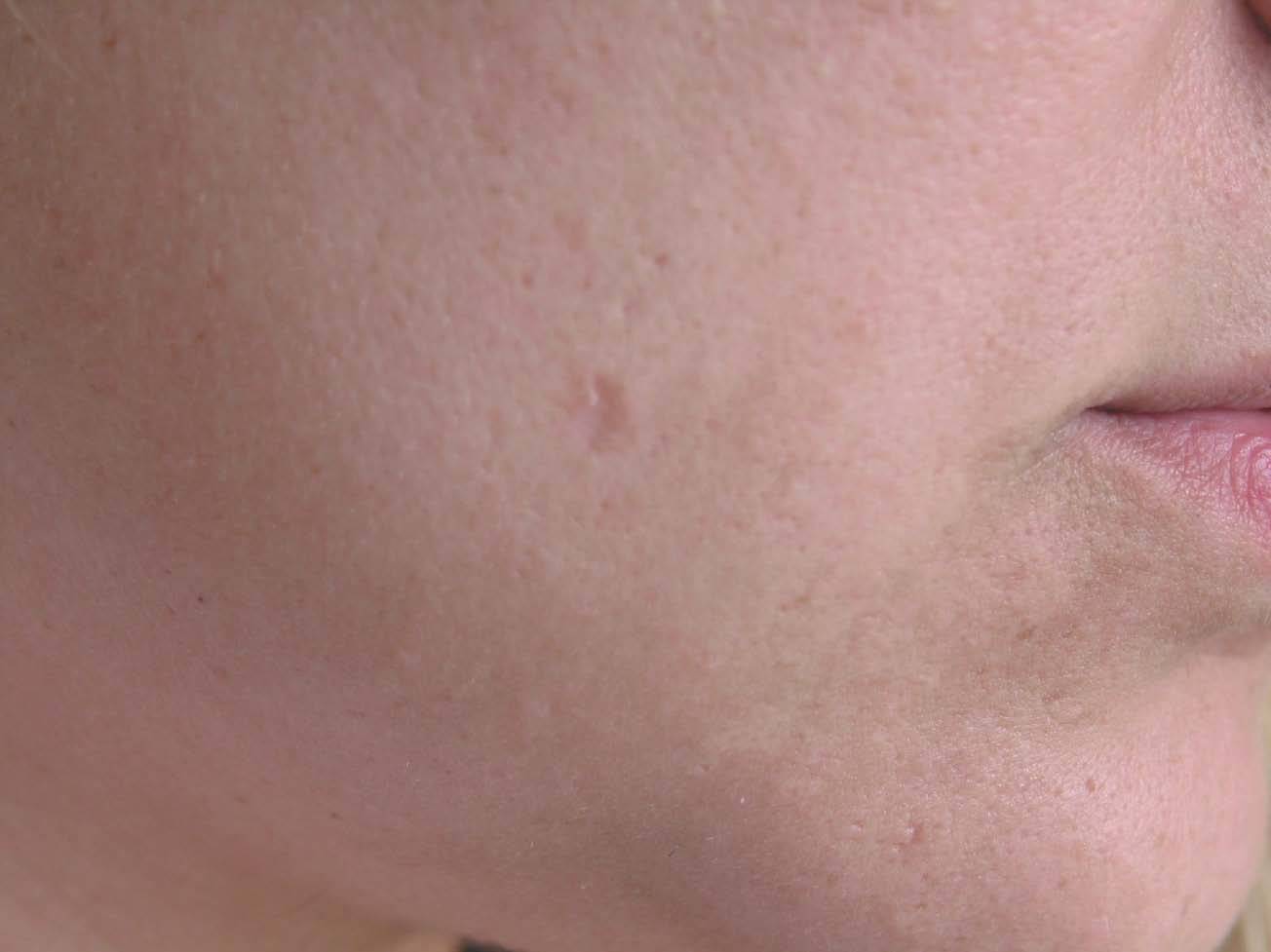
Fig. 1.--Lesión anular única de bordes elevados y centro ligeramente deprimido, localizada en la mejilla derecha, próxima a la comisura bucal correspondiente a la paciente del caso clínico 1.
Caso 2
Una mujer de 30 años de edad, sin antecedentes patológicos de interés, salvo apendicectomía en la infancia y en tratamiento actual con anticonceptivos orales, acudió a nuestra consulta para la valoración de una lesión cutánea asintomática de 2 años de evolución, que había aumentado lentamente de tamaño y para la cual no había recibido tratamiento.
A la exploración física se observaba en la región supralabial derecha, una única lesión papulosa anular, de coloración similar a la piel, de bordes elevados y centro ligeramente deprimido, no adherida a planos profundos, de 7 mm de diámetro (fig. 2). No se palparon adenopatías locorregionales y el resto de la exploración cutánea fue normal.
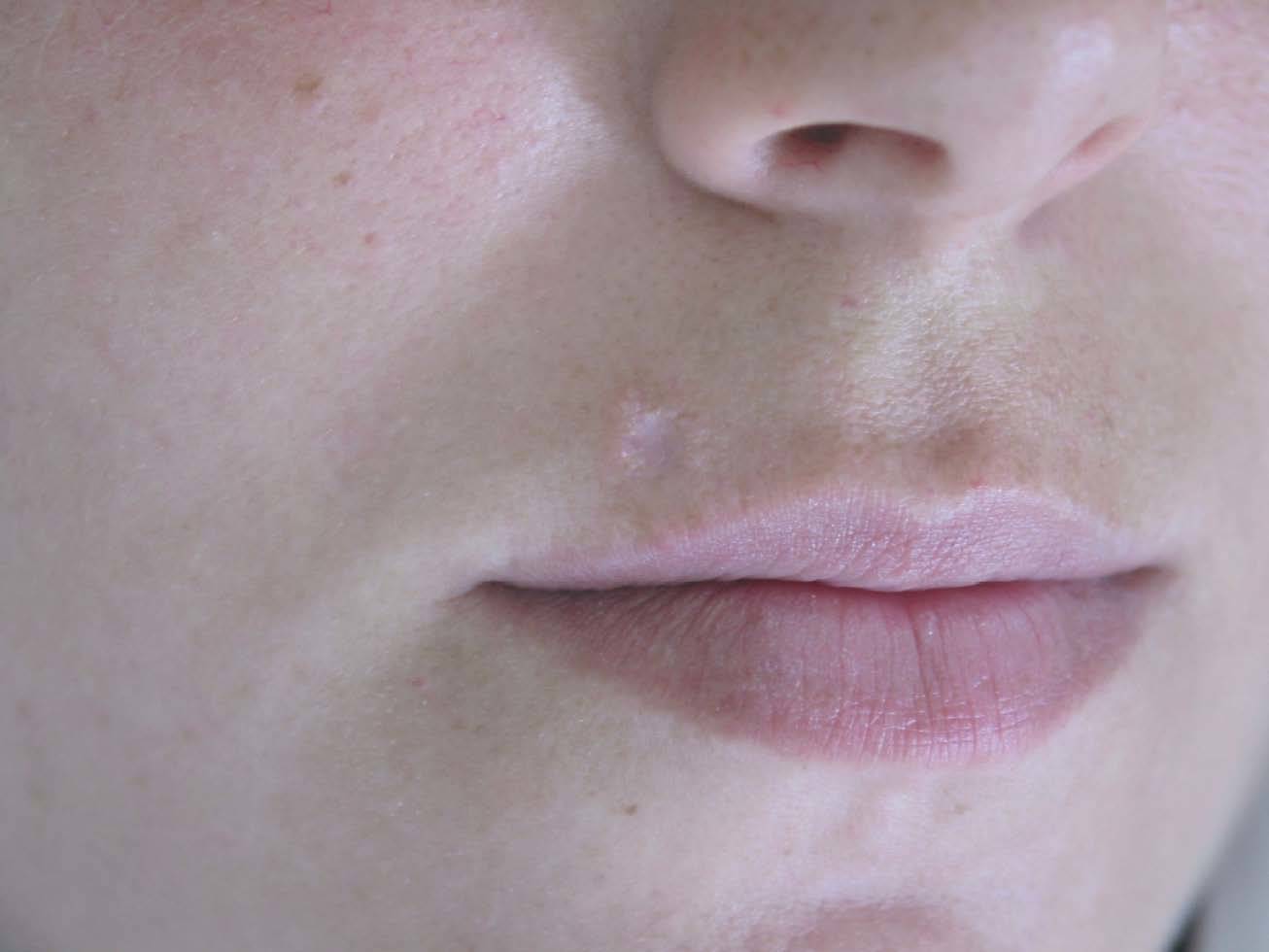
Fig. 2.--Lesión papulosa anular, de coloración similar a la piel, de bordes elevados y de centro ligeramente deprimido, localizada en la región supralabial derecha, correspondiente a la paciente del caso clínico 2.
El estudio histopatológico de ambas lesiones fue superponible totalmente a las 2 pacientes, apreciándose una tumoración dérmica, no encapsulada, compuesta por un estroma desmoplásico que ocupa la parte superior de la dermis (fig. 3) y por cordones y nidos de células basaloides. También se pueden apreciar quistes infundibulares rellenos de queratina hojaldrada basófila entremezclados con los cordones basaloides (fig. 4). Rodeando a estos cordones y a los nidos epiteliales se pueden apreciar anillos de colágeno eosinófilo y esclerótico (fig. 5).
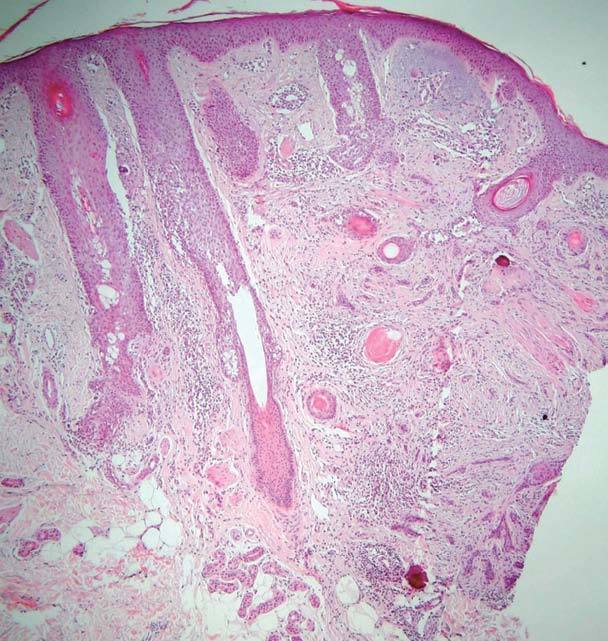
Fig. 3.--Presencia de pequeños islotes y cordones de células basaloides con escaso citoplasma sobre un estroma fibroso, localizados en la dermis superior y media. (Hematoxilina-eosina, x4.)
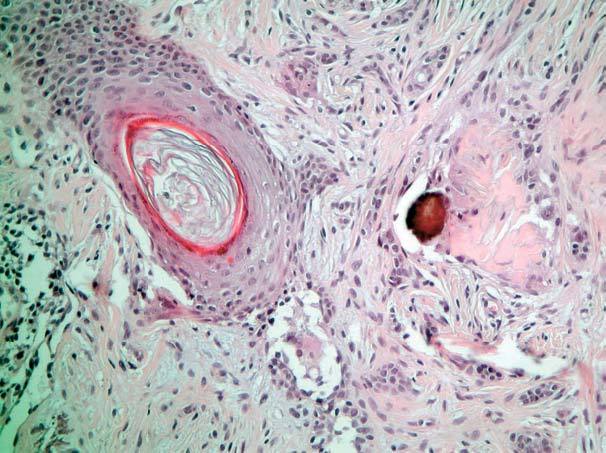
Fig. 4.--Detalle de los pequeños quistes infundibulares presentes en las áreas más superficiales de la dermis. (Hematoxilina-eosina, x10.)
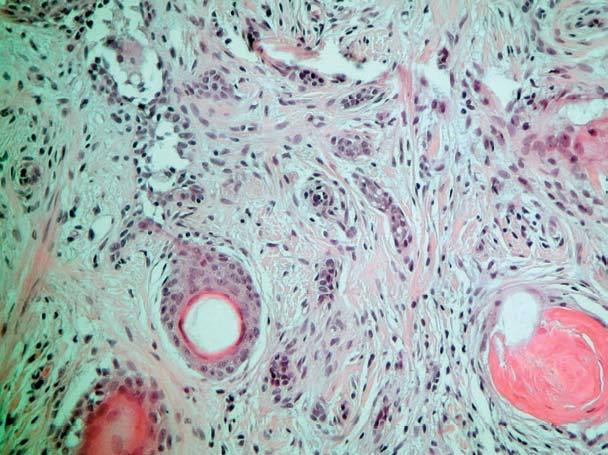
Fig. 5.--Presencia de quistes infundibulares rellenos de queratina que se encuentran entremezclados con cordones de epitelio basaloide. Muchos de estos cordones epiteliales están rodeados de anillos eosinófilos de colágeno esclerosado. (Hematoxilina-eosina, x20.)
DISCUSION
El primer autor que describió una neoplasia compatible clínicamente con el TD fue Hartzell en 1904 1. Posteriormente de forma sucesiva se fueron describiendo varios casos de TD con diferentes términos; en 1909, Schopper 2 lo denominó «epitelioma adenoide quístico», Ingels 3 en 1935 «epitelioma adenoide quístico con hallazgos de siringoma», Zeligman 4 en 1960 «tricoepitelioma solitario» y en 1969 Pinkus y Mehregan 5«morphea-like epithelioma». Bajo estas diferentes denominaciones podemos encontrarnos en la bibliografía al TD, término que acuñado por Brownstein y Shaphiro 6 en 1977, para describir esta neoplasia específica y diferenciarla de otras neoplasias con diferenciación folicular. En ese mismo año y de forma independiente, MacDonald et al 7, describieron el TD como una neoplasia fibroepitelial benigna, denominada «hamartoma epitelial esclerosante».
Desde el punto de vista clínico, el TD se presenta como una lesión papulosa anular de pequeño tamaño (generalmente < 1 cm), de coloración blanco-amarillenta, indurada, de consistencia firme y con un borde elevado y centro deprimido. Generalmente es solitaria y se localiza en la cara, con predominio en las mejillas de mujeres jóvenes (85 %), cerca de la comisura labial y excepcionalmente puede aparecer en el cuero cabelludo, en el cuello y en la parte superior del tronco 6. Se han descrito casos de afectación múltiple, y aunque la mayoría de los casos son asilados también existen formas familiares 8,9. Suele aparecer alrededor de la tercera década de la vida y el diagnóstico se demora a veces durante varios años. De forma excepcional se han descrito casos en los que la infiltración por TD en la cara, simulaba una lepra lepromatosa 10.
Histopatológicamente se caracteriza por ser una lesión simétrica, bien circunscrita y limitada a la dermis superficial y media, extendiéndose de forma muy rara al tejido celular subcutáneo. Dicha lesión está constituida por cordones y nidos de células basaloides que pueden conectar con la epidermis y con los infundíbulos foliculares preexistentes. Entre estos cordones basaloides se pueden observar entremezclados pequeños quistes infundibulares, que contienen queratina hojaldrada y ortoqueratósica. Puede observarse una reacción granulomatosa de cuerpo extraño por rotura de estos quistes, o una calcificación de los mismos.
El diagnóstico del TD es en ocasiones difícil, pues precisa de un amplio diagnóstico diferencial clínico e histológico con otras entidades frecuentes en la clínica diaria. Desde el punto de vista clínico debe distinguirse del carcinoma basocelular morfeiforme (CBCM), de la hiperplasia sebácea, del tricoepitelioma solitario, del granuloma anular o de la esclerodermia 6. Histológicamente comparte ciertas características también con el CBCM, con el cual a veces es muy difícil de diferenciar, con el siringoma, carcinoma anexial microquístico, hamartoma folicular basaloide y con el tricoadenoma 11. Realmente el dilema diagnóstico se plantea entre el TD y el CBCM, tanto clínica como histológicamente. Hay datos a favor del TD, desde el punto de vista clínico aparece en mujeres jóvenes y no se ulcera en el centro, y desde el punto de vista histológico, se observa cómo la presencia de células tumorales están limitadas al folículo del pelo formando una neoplasia simétrica, bien circunscrita y superficial, sin penetrar más allá de la dermis media, con islotes epiteliales pequeños y uniformes, quistes córneos y con un estroma desmoplásico 6,12,13. Un hallazgo característico del TD, y que es muy útil para el diagnóstico diferencial con otros tumores, en especial con el CBCM, es la presencia de anillos de colágeno eosinófilo y esclerótico rodeando los cordones y los nidos epiteliales de la neoplasia 14,15.
Pueden emplearse las técnicas inmunohistoquímicas con estromelisina-3 y con antígeno carcinoembrionario, para completar el diagnóstico; siendo ambos estudios negativos para el TD, que apoyan su origen pilar 13,16. Además el TD puede expresar citoqueratinas asociadas a la vaina radicular externa, como la CK-15 que se utiliza como marcador diferencial con el CBC (en cuyo caso siempre son negativas) 17.
Aproximadamente en el 15 % de los TD se encuentran tecas o nidos de un nevo melanocítico intradérmico entremezclados con los cordones epiteliales correspondientes al TD, por ello esta lesión se considera más un hamartoma que una neoplasia 14,18,19.
Hemos descrito 2 pacientes diagnosticadas de TD; su importancia radica en la similitud clínica e histológica con el CBC, siendo el estudio histopatológico indispensable para llegar a un diagnóstico definitivo. Este diagnóstico es imprescindible dado que el pronóstico y el tratamiento de ambas entidades es muy diferente.
Correspondencia:
M.ª Teresa Bordel. Martínez Villergas, 6, 1.º-B.
49003 Zamora. España.
maitebordel@aedv.es
Recibido el 25 de noviembre de 2004.
Aceptado el 24 de marzo de 2006.


