




INTRODUCCION
El síndrome de Touraine-Solente-Golé, paquidermoperiostosis u osteoartropatía hipertrófica primaria, descrito inicialmente en 1868 por Friedreich1, es una enfermedad hereditaria infrecuente que afecta a la piel, al tejido conjuntivo, a los huesos, a las articulaciones y al sistema nervioso autónomo. Se presenta con mayor frecuencia en varones que en mujeres (9:1) y su inicio es bimodal, con un pico de incidencia en el primer año de vida y el segundo a los 15 años2. La osteoartropatía hipertrófica (OAH) puede presentarse como una forma primaria o síndrome de Touraine-Solente-Golé3 y una forma secundaria asociada a procesos neoplásicos. Se presenta un nuevo caso de esta entidad, en el que el motivo de consulta fue una proliferación de partes blandas en las piernas, asociada a lesiones de dermatitis de estasis.
DESCRIPCION DEL CASO
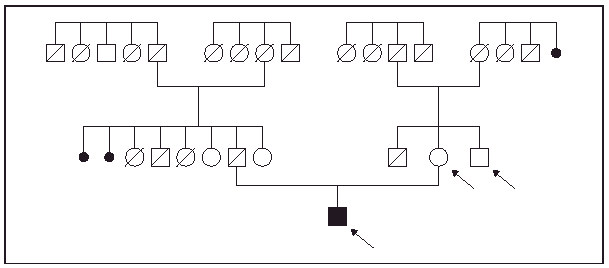
Un varón de 47 años de edad fue remitido a nuestro servicio para la valoración de unas lesiones excrecentes en el tercio distal de las piernas que habían ido creciendo de forma progresiva durante los últimos 8 años. Entre sus antecedentes personales destacaban una hipercolesterolemia controlada con dieta y una litiasis biliar asintomática. Entre los antecedentes familiares figuraba la existencia de consanguinidad: una tatarabuela por parte de padre y un bisabuelo por parte de madre eran hermanos.
En la exploración física presentaba un engrosamiento cutáneo, de aspecto tumoral, en la mitad inferior de ambas piernas. En la pierna izquierda se observaba una tumoración, adherida a planos profundos de consistencia variable, entre blanda y firme según la zona, de 15 ×9 cm de diámetro localizada en la cara interna del tobillo izquierdo. La superficie tumoral era del color de la piel normal, hiperhidrótica y en la zona superoposterior presentaba una placa violácea, de superficie irregular, bien delimitada. La lesión de la pierna derecha presentaba características similares, pero de menor tamaño. En la exploración física general el paciente presentaba unos rasgos fenotípicos distintivos: un aspecto acromegálico, la piel de la cara engrosada, grasa y con los surcos prominentes, los dedos de las manos en palillo de tambor y las uñas «en vidrio de reloj», una hiperhidrosis palmoplantar y, en los pies, presentaba hallux valgus muy marcado junto con hiperqueratosis plantar. Durante la anamnesis el paciente refirió que ningún familiar próximo presentaba ninguna de estas manifestaciones y en exploración física de la madre y el tío materno no se observaron manifestaciones similares. Asimismo, el paciente nos trajo fotografías de la familia paterna y tampoco se detectaron ninguno de los hallazgos presentados por el paciente (figs. 1-5).
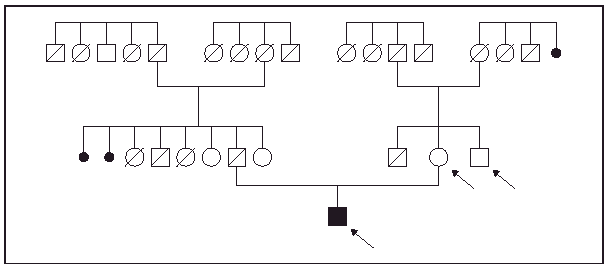
Fig. 1.--Árbol genealógico del paciente. Las flechas indican los casos en los que se realizó una minuciosa exploración física.
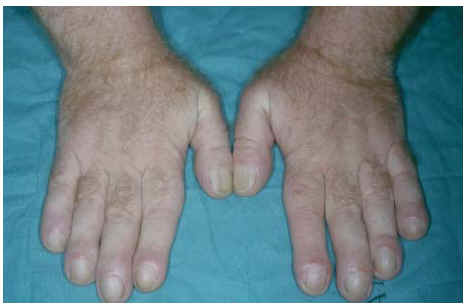
Fig. 2.--Manos con dedos «en palillo de tambor» y uñas «en vidrio de reloj».
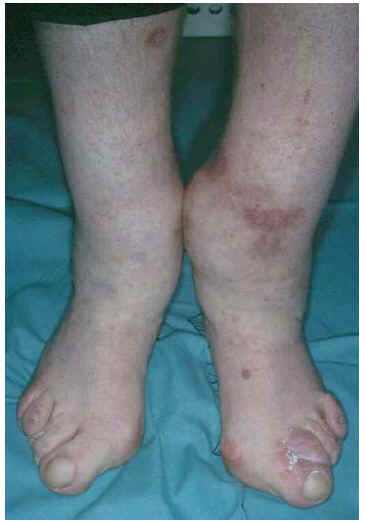
Fig. 3.--Ensanchamiento de la porción distal de las extremidades inferiores, placa eritematosa característica de la dermatitis de estasis.
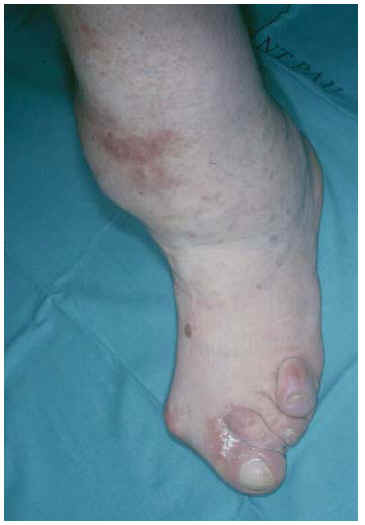
Fig. 4.--Lesión tumoral de color piel normal con mácula eritematosa en tercio inferior de pierna izquierda.
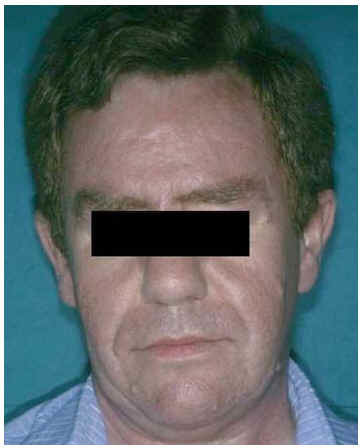
Fig. 5.--Facies con piel gruesa, grasa, eritematosa y brillante.
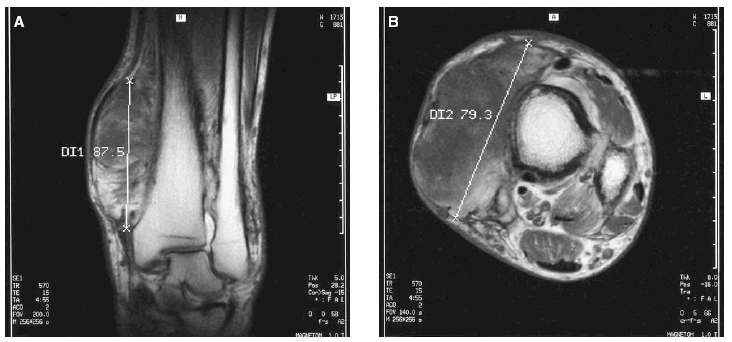
En la analítica sanguínea no se observaron alteraciones bioquímicas ni en el hemograma. Los niveles séricos de testosterona, cortisol, tirotropina, prolactina, lutropina, folitropina y hormona del crecimiento se hallaron dentro de los valores de referencia. El examen histopatológico de una biopsia «en sacabocados» de la lesión violácea del tobillo izquierdo puso de manifiesto una dermatitis de estasis. En el estudio mediante resonancia magnética (RM) de las extremidades inferiores se observó un engrosamiento irregular de la cortical con algunas imágenes de espiculación y con un ensanchamiento metafisario en ambas piernas. La afectación de partes blandas era bilateral, localizada en el tercio distal, más prominente en el lado izquierdo y consistía en una proliferación seudotumoral del tejido adiposo que se infiltraba entre los músculos hasta alcanzar planos yuxtaarticulares asociado a cambios de fibrosis (fig. 6). Se realizó una tomografía computarizada (TC) toracoabdominal que fue normal y que descartó patología pleuropulmonar, hepática y gastrointestinal.
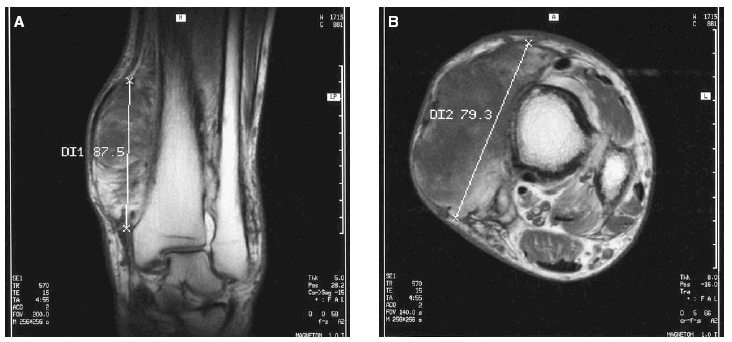
Fig. 6A y B.--Proliferación seudotumoral del tejido adiposo que se infiltra entre los músculos asociado a cambios de fibrosis en el estudio mediante RM.
Ante los hallazgos clínicos radiológicos, y tras descartar que se tratara de una forma secundaria de OAH, se concluyó que se trataba de un síndrome de Touraine-Solente-Golé y se inició tratamiento sintomático para la dermatitis de estasis con medidas posturales, vendas elásticas y venotónicos.
COMENTARIO
El síndrome de Touraine-Solente-Golé, paquidermoperiostosis u OAH primaria fue descrito inicialmente en 1868 por Friedreich1 en dos hermanos varones jóvenes, aunque el nombre de síndrome de Touraine-Solente-Golé3 le fue otorgado en 1935 por estos autores que reconocieron que se trataba de una enfermedad familiar con tres formas de presentación: una forma completa, que cursaba con paquidermia y periostosis; una forma incompleta, sin la existencia de paquidermia, y una forma mínima, en la que existía paquidermia con mínimos cambios óseos. Solamente representa del 3 al 5 %4 de las formas de OAH debido a que la mayoría de los pacientes presentan una forma de OAH secundaria, asociada a enfermedades benignas o tumorales de origen cardiopulmonar, endocrinológico, gastrointestinal, u otros2.
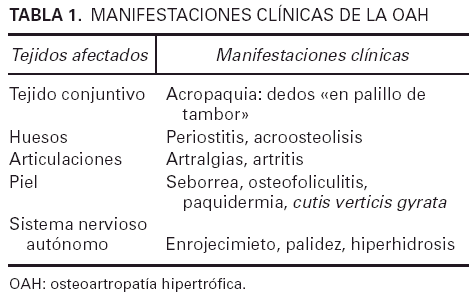
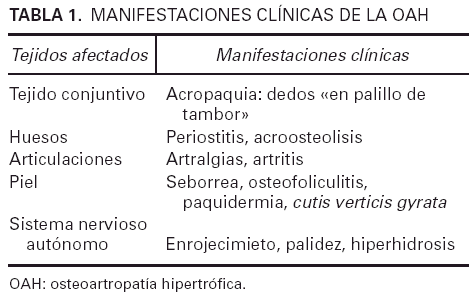
Las manifestaciones clínicas de la enfermedad dependen del tejido afectado pero las alteraciones del tejido conjuntivo, los huesos y la piel son las más habituales (tabla 1). Entre ellas destacan la acropaquia y el engrosamiento cutáneo y del periostio de la parte distal de las extremidades, así como la seborrea y la hiperhidrosis. La expresión clínica más relevante es la acropaquia bilateral de manos y pies, que consiste en un aumento de la convexidad de las uñas y de los lechos ungueales, tanto en sentido longitudinal como transversal. El ángulo de Lovibond es mayor de 180° y el ángulo de Curth es inferior a 160°, debido a una hipertrofia de los tejidos blandos en los pulpejos digitales. Además, la lámina ungueal es dura por hiperplasia del tejido fibrovascular de la base de la uña5. Las causas de acropaquia con las que hay que realizar el diagnóstico diferencial son numerosas y entre ellas se incluyen los rasgos raciales, que se trate de un defecto genético aislado, la policitemia primaria o secundaria, la intoxicación por fósforo, arsénico o mercurio, la subluxación del hombro con parálisis del plexo braquial, la enfermedad de Graves, etc.5. Otro hallazgo característico es el engrosamiento cutáneo o «paquidermia» que afecta la piel de la cara, el cuero cabelludo y la parte superior del tronco. Los pliegues de la frente y los pliegues nasolabiales suelen estar muy marcados. Esta facies típica se denomina cutis verticis gyrata. En los párpados el engrosamiento puede ocasionar una ptosis mecánica. La piel es de color pardusco, lustrosa y brillante tanto por la importante secreción sebácea como por la excesiva sudoración. En algunos casos se puede observar osteofoliculitis e hiperplasia de glándulas sebáceas4,6.
El compromiso óseo se manifiesta por un aumento de tamaño de las extremidades que se asemeja clínicamente a la acromegalia. Lo más característico es un ensanchamiento de la porción distal de las extremidades inferiores, con aspecto elefantiásico, debido a la proliferación de los huesos tubulares. Esto es consecuencia de una reacción inflamatoria en el periostio, seguida de un engrosamiento por aposición de hueso nuevo, con disposición laminada en imagen «en cebolla», con conservación del espacio articular, sin erosión ni osteopenia paraarticular. El proceso comienza de forma prominente en el periostio y se extiende a partir de la diáfisis y metáfisis hasta la epífisis4,7. Estos cambios radiográficos con reacción perióstica también pueden encontrarse en otras enfermedades como la sífilis, el escorbuto, o las intoxicaciones por estroncio, flúor, vitamina A y D. Otros hallazgos descritos en el síndrome de Touraine-Solente-Golé, pero no observados en nuestro caso, son las calcificaciones ligamentosas, la acroosteolisis, la afectación raquídea y la displasia epifisaria múltiple.
La etiología de esta entidad aún se desconoce. Aunque inicialmente se habló de una transmisión genética autosómica dominante con expresividad variable3,4,8, el considerable número de casos de hermanos afectados con padres aparentemente sanos y varios casos con antecedentes de consanguinidad, como el nuestro, sugieren un patrón de herencia recesivo9-11. También se han descrito casos relacionados con anomalías cromosómicas como la trisomía XYY4. Debido a la similitud de las manifestaciones clínicas de la osteoartropatía hipertrófica primaria y secundaria se piensa que el mecanismo patogénico sería el mismo.
Se han descrito numerosas complicaciones evolutivas en este proceso patológico, como la ptosis palpebral que muchas veces requiere tratamiento quirúrgico, alteraciones auditivas, cifosis, radiculopatías y artrosis. Genéticamente suele asociarse con endocrinopatías, úlceras duodenales y gastritis hipertrófica que modifican o empeoran el pronóstico. En general la esperanza de vida suele ser normal. El tratamiento es sintomático y de las posibles complicaciones que pudieran presentarse.
El paciente que se presenta hasta el momento no ha presentado patologías relacionadas genéticamente con la OAH primaria, no manifiesta una excesiva preocupación por sus rasgos acromegálicos y no se han detectado complicaciones evolutivas que alteren su calidad de vida, motivo por el que se instauró tratamiento sintomático para la dermatitis de estasis.
El interés del cuadro radica en su expresividad clínica variable y en la importancia de determinar si se trata de una OAH primaria o secundaria. Las manifestaciones clínicas son similares y sólo puede diferir el momento de su aparición. Es importante recordar que la forma primaria tiene una evolución crónica con complicaciones evolutivas, pero la esperanza de vida suele ser normal, mientras que las formas secundarias se asocian a neoplasias, especialmente de origen pulmonar, preceden al diagnóstico de la misma varios meses y tras el tratamiento de la patología tumoral hay una involución de los signos y síntomas de la OAH. Por todo ello es importante tener en cuenta que es necesario realizar un estudio exhaustivo de estos pacientes para obtener de forma temprana un diagnóstico preciso.


