



INTRODUCCION
Los anticuerpos antifosfolípidos (APL) son un grupo de autoanticuerpos dirigidos contra una amplia gama de fosfolípidos que tienen carga negativa. Su presencia en suero se ha relacionado con diversos trastornos vasculares que genéricamente se conocen como síndrome de anticuerpos APL.
La vasculitis necrosante es una de las manifestaciones cutáneas del síndrome de anticuerpos APL, que puede llegar a afectar del 20 al 30 % de los casos1,2.
DESCRIPCION DEL CASO
Una mujer mexicana de 52 años de edad, presentaba una dermatosis que afectaba al pabellón auricular derecho a nivel del hélix, extremidad superior izquierda a nivel de codo y articulación metacarpofalángica, extremidades inferiores en dorso de pie izquierdo y segundos dedos de pies de forma bilateral. En la exploración se observaron úlceras de 1 a 6 cm, algunas cubiertas por escaras y otras con fondo sucio por la presencia de exudado seropurulento escaso, con bordes bien definidos y eritema periférico en algunas de ellas (figs. 1 y 2). Las lesiones se iniciaron por un eritema que aduccionó hacia la formación de úlceras que cursaron asintomáticas. La paciente fue hospitalizada, documentándose anemia microcítica hipocrómica, 273.000 plaquetas; test de serología luética, virus de la inmunodeficiencia humana (VIH), serología para hepatitis B y C, anticuerpos antinucleares negativos, anticardiolipinas (ACL) positivas, crioglobulinas positivas, beta 2-glucoproteína 1 (β2GP1) positiva y electroforesis de proteínas que mostró una gammapatía policlonal.
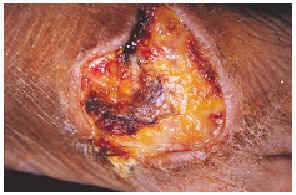
Fig. 1.--Úlcera en el dorso del pie derecho.
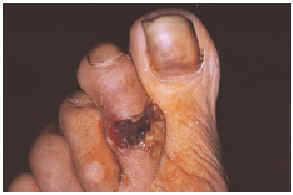
Fig 2.--Lesión ulcerosa en el dorso del segundo dedo del pie derecho.
El cultivo de piel no fue relevante. El estudio histopatológico de piel mostró necrosis dermoepidérmica, y vasos congestivos y engrosados, así como la presencia de trombos en vías de resolución en pequeños vasos de la dermis (fig. 3). La inmunofluorescencia directa positiva para inmunoglobulinas, fibrinógeno y C3 en la pared de vasos de pequeño calibre de la dermis fue negativa. Una telerradiografía de tórax mostró cardiomegalia grado I y abombamiento de arco pulmonar. La gammagrafía pulmonar perfusoria con hipoperfusión leve y un ecocardiograma pone de manifiesto hipertensión pulmonar. Las pruebas de función respiratoria dieron como resultado una moderada restricción de la capacidad vital. Con estos datos se llegó al diagnóstico de hipertensión pulmonar.
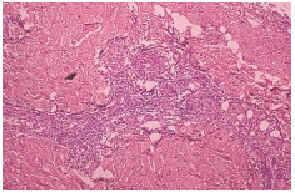
Fig. 3.--Trombosis de pequeños vasos en la dermis.
La paciente fue tratada con 1 mg/kg/día de prednisona y 7,5 mg de metotrexato semanalmente, 100 mg diarios de antiagregante plaquetario, 800 mg diarios de pentoxifilina y suplementos con hierro. Se aplicaron tópicamente apósitos hidrocoloides y cuidados específicos de las úlceras, con lo que la respuesta fue satisfactoria, obteniéndose remisión total de las lesiones a los 4 meses.
DISCUSION
El síndrome de anticuerpos APL es un estado protrombótico adquirido y autoinmune de hipercoagulación sistémica en el cual hay trombosis a diferentes niveles. Los criterios principales comprenden trombosis arterial o venosa, pérdida fetal recurrente y trombocitopenia asociada a la presencia de anticoagulante lúpico (AL) o elevación de los anticuerpos ACL.
Este síndrome se ha dividido en primario y secundario. El primero ocurre cuando hay ausencia de otra enfermedad conocida o asociada y es más frecuente que el secundario. Éste se refiere a la presencia del síndrome asociado a otras enfermedades, especialmente inmunológicas, como el lupus eritematoso, el síndrome de Sjögren, esclerodermia, crioglobulinemia, enfermedades linfoproliferativas, enfermedades infecciosas como sífilis, infección por VIH, hepatitis C o borreliosis, y enfermedades asociadas al uso de medicamentos como clorpromacina, hidralacina, fenitoína e interferón1,4.
Para el diagnóstico del síndrome de anticuerpos APL existen unos criterios mayores que son trombosis venosa o arterial, pérdida fetal recurrente y trombocitopenia, y unos criterios serológicos que son la presencia de ACL, AL y β2GP1.
Se considera diagnóstico del síndrome la presencia de un criterio mayor y uno serológico, pero este último tendrá que ser positivo en por lo menos dos ocasiones en un lapso medio de tiempo de 8 semanas5.
Estudios previos publicados4,6,7, señalan que del 2 al 8 % de la población normal tiene anticuerpos APL detectables sin presentar enfermedad clínica, y otros estudios afirman que el 41 % de los pacientes con AL tienen lesiones en piel como primera manifestación de la enfermedad, la mayoría de los cuales se relacionan con oclusión vascular demostrable por hallazgos histopatológicos4,6-8. No es frecuente encontrar presentes los dos anticuerpos en un mismo sujeto, de manera que, dependiendo del anticuerpo presente, diversos autores denominan al síndrome APL-ACL o APL-AL; el primero de ellos se asocia con más frecuencia a trombosis arteriales y el segundo a trombosis venosas.
Los anticuerpos ACL se encuentran más frecuentemente que el AL en proporción 5:1 y predominan en personas mayores. Habitualmente son isotipos IgG, especialmente subclases IgG2 e IgG4 y requieren de la presencia de un cofactor que es la β2GP1 también llamada apolipoproteína H9. Los anticuerpos ACL tienen propiedades procoagulantes, ya que inhiben la vía intrínseca de la coagulación. Pueden encontrarse ACL positivos en otras enfermedades mientras que la detección de la β2GP1 se considera específica y confirmatoria del síndrome de anticuerpos APL.
El AL, identifica a una familia de anticuerpos APL presentes en el suero, generalmente del tipo IgG y ocasionalmente IgM o IgA o ambas, que interfieren con las reacciones de coagulación que dependen de fosfolípidos. Debe sospecharse la presencia de AL al encontrar un tiempo parcial de tromboplastina alargado; si este parámetro de laboratorio o cualquiera de los tiempos de coagulación no se corrige al agregar plasma normal, se considera positivo para AL10. El AL se encuentra más frecuentemente en mujeres jóvenes.
Los hallazgos histopatológicos de este síndrome consisten en trombosis no inflamatorias de vasos pequeños de la dermis como hallazgo más común, que pueden acompañarse de proliferación capilar en dermis subpapilar. Los hallazgos de inmunofluorescencia directa incluyen depósitos de fibrinógeno, IgM granular y C3 en los vasos dérmicos7-11.
El tratamiento y la profilaxis se basan principalmente en el uso de anticoagulantes y agentes antiplaquetarios, junto con la utilización de prednisona, agentes inmunosupresores, pulsos de ciclofosfamida, plasmaféresis e infusiones con gammaglobulinas.
Para la resolución de las úlceras se recomiendan la utilización tópica de antisépticos locales, apósitos hidrocoloides o de alginato; en otros casos puede recurrirse a la desbridación quirúrgica y, en casos extremos, la realización de colgajos o la aplicación de injertos11.


