






INTRODUCCION
Los primeros casos descritos que se refieren a la profusión de queratosis seborreicas como fenómeno paraneoplásico datan de 1890, y se deben, de forma independiente, al francés Ulysse Trélat y al alemán Edmund Leser1. Sin embargo, hasta 1965 no se acuño el término «signo de Leser-Trélat» para denominar la entidad descrita por estos autores.
Años más tarde, Rampen y Schwengle cuestionaron los trabajos de Leser y Trélat, alegando que los pacientes descritos por ambos presentaban angiomas seniles y no queratosis seborreicas2. Estos autores atribuían a Höllander la primera descripción de esta dermatosis paraneoplásica. A pesar de ello, se han publicado nuevos casos posteriormente y en todos ellos se mantiene el epónimo inicial de signo de Leser-Trélat.
DESCRIPCION DEL CASO
Un varón de 82 años, con antecedentes personales de etilismo crónico, consultó por la aparición en los 6 últimos meses de múltiples lesiones verrucosas, pruriginosas, en tronco, cara y raíz de miembros. No refería otros síntomas, salvo la pérdida de 2 kg de peso en el último mes. A la exploración el paciente presentaba una eritrodermia generalizada, acompañada de numerosas lesiones verrucosas pigmentadas, de entre 1 y 6 cm de tamaño, de tacto untuoso, en las localizaciones antes mencionadas (fig. 1). También se detectaron múltiple adenopatías rodaderas, no dolorosas, algunas de gran tamaño, en regiones axilares e inguinales. El resto de la exploración física fue normal.
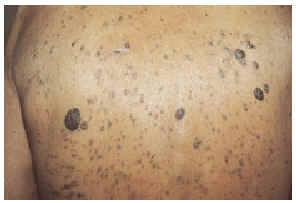
Fig. 1.--Erupción de múltiples queratosis seborreicas, de entre 1 y 6 cm de tamaño, en tronco.
Una biopsia de la piel eritrodérmica mostraba un intenso infiltrado linfocitario pleomórfico, en banda subepidérmica, con epidermotropismo, así como fenómenos dispersos de exocitosis linfocitaria y microabscesos de Pautrier (figs. 2 y 3): el infiltrado linfocitario era positivo para CD4 y CD3, y negativo para CD22 y CD8 (fig. 4). El estudio histológico de una adenopatía mostró una infiltración específica por linfoma.
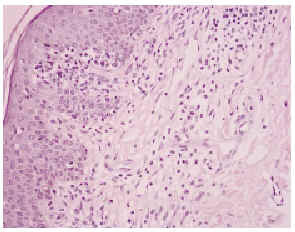
Fig. 2.--Infiltrado linfocitario subepidérmico, con epidermotropismo y fenómenos de exocitosis linfocitaria.
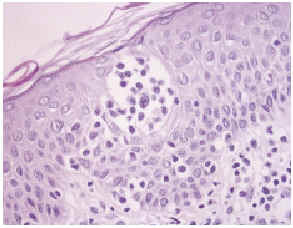
Fig. 3.--Detalle de un microabsceso de Pautrier.
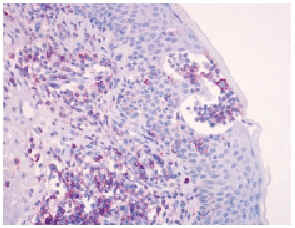
Fig. 4.--Positividad para la tinción con CD4 y detalle de microabsceso de Pautrier.
La biopsia de una de las lesiones cutáneas verrucosas resultó compatible con el diagnóstico de queratosis seborreica (fig. 5), junto con un infiltrado linfocitario similar al observado en la biopsia de la piel eritrodérmica.
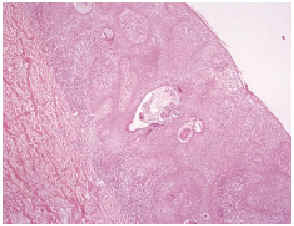
Fig. 5.--Coexistencia de imágenes histológicas de queratosis sebo rreica con infiltrado linfocitario epidermotropo.
En el hemograma destacaba una leucocitosis de 19,6 ×109/l, con presencia de un 23 % de células de Sézary. En la punción-aspiración de médula ósea, se evidenciaba una discreta infiltración de alrededor del 7 % por células de Sézary, que en el estudio inmunohistoquímico mostraron marcadores compatibles con el proceso linfoproliferativo detectado en piel y sangre periférica. Sin embargo, en la biopsia de médula ósea no se hallaron alteraciones significativas.
En las pruebas de imagen realizadas, incluyendo enema opaco, rectosigmoidoscopia, ecografía abdominal y tomografía computarizada (TC) toracoabdominal no se detectaron hallazgos relevantes, salvo la confirmación de la existencia de adenopatías axilares e inguinales de hasta 1,5 cm, mediante la TC.
Con estos datos, se llegó al diagnóstico de signo de Leser-Trélat asociado a síndrome de Sézary. Se inició tratamiento con interferón α2a, a dosis de 3 × 106 UI, con posterior elevación a 6 ×106 UI, asociado a psoraleno y luz ultravioleta (PUVA), observándose inicialmente una mejoría clínica e histológica de la neoplasia, así como una considerable reducción en el número de queratosis seborreicas.
Tras 3 meses de tratamiento el paciente se negó a continuar la terapia. Fue ingresado en varias ocasiones por diversos cuadros infecciosos, y falleció al año de iniciarse el proceso por empeoramiento.
DISCUSION
El signo de Leser-Trélat se caracteriza por la erupción brusca de numerosas queratosis seborreicas, con frecuencia pruriginosa, secundaria a una neoplasia. La existencia del signo de Leser-Trélat ha sido objeto de debate en los últimos años. Tanto las queratosis seborreicas como la aparición de neoplasias internas son muy frecuentes en los ancianos, por lo que la coexistencia de ambos cuadros es bastante usual. Revisiones de la literatura médica1,2 apuntan a que muchos de los casos publicados están pobremente documentados y presentan factores de confusión como la consideración de cualquier tipo de lesión verrucosa como queratosis seborreicas o la consideración como signo de Leser-Trélat de cuadros que cursan con profusión de verrugas seborreicas en el curso de dermatosis inflamatorias por lo que el carácter paraneoplásico de la sintomatología en estos casos es dudoso.
En la actualidad, para muchos autores, el signo de Leser-Trélat constituye un proceso neoplásico facultativo, ya que en ocasiones la profusión de queratosis seborreicas puede estar desencadenada por entidades como el embarazo3 o aparecer en el curso de dermatoris inflamatorias como eritrodermias, eccemas o lepra lepromatosa4-6. Otros cuadros dermatológicos que se han asociado a este signo son la acantosis nigricans y la hiperqueratosis palmoplantar7-9.
Entre las neoplasias asociadas con mayor frecuencia destacan con un 55 % adenocarcinomas gástrico y de colon seguidos de los tumores hematológicos con un 20 %5,10-12, sobre todo neoplasias de la serie linfoide. El signo de Leser-Trélat es muy infrecuente, incluso en pacientes con cáncer. En la revisión bibliográfica más reciente se recoge un total de 91 casos publicados5. La asociación con el síndrome de Sézary es rara, hasta el momento se reduce a cinco el número de casos descritos1,10,11,13-15 (tabla 1).

La profusión de queratosis seborreicas puede afectar a cualquier localización cutánea, pero la localización más frecuente es el tronco, seguido de las extremidades. El concepto del signo de Leser-Trélat implica la aparición brusca de las lesiones cutáneas; se estima que el tiempo medio de instauración de las lesiones dermatológicas es de alrededor de 15 semanas7. En algunos trabajos se ha observado una relación inversamente proporcional entre la edad del paciente y el tiempo de instauración del cuadro cutáneo2. El síntoma asociado con mayor frecuencia es el prurito, presente en alrededor del 40 % de los pacientes2,5,7,9. En ocasiones, es difícil diferenciar si este síntoma es secundario a la dermatosis paraneoplásica o bien al proceso subyacente, como ocurre en nuestro caso. En la mayoría de los casos publicados la aparición de las lesiones cutáneas y del tumor suele ser simultánea, si bien de forma paradójica sólo en un tercio de los pacientes se describe un curso paralelo entre ambos procesos2,7,9. Nuestro paciente, al igual que 4 de los 5 casos de síndrome de Sézary asociados al signo de Leser-Trélat hallados en la literatura médica1,10,11,13-15, presentó una evolución paralela entre la sintomatología cutánea y la hematológica, con desaparición parcial de las queratosis seborreicas a medida que la neoplasia respondía al tratamiento específico con PUVA e interferón-α).
Otro punto criticado en algunas revisiones2 ha sido la confusión clínica entre queratosis seborreicas y otros tipos de lesiones verrucosas, debido a la falta de confirmación histológica de las lesiones cutáneas. Sólo en uno de los 5 casos descritos de Leser-Trélat asociado a síndrome de Sézary se realizó biopsia cutánea para confirmar el diagnóstico de queratosis seborreica11. En dicha biopsia, al igual que en nuestro caso, coexistían imágenes histológicas de verruga seborreica y de linfoma de células T.
Existen varias teorías patogénicas sobre la aparición del signo de Leser-Trélat. La más aceptada apunta a la producción por parte del tumor de diversos factores de crecimiento, habiéndose observado una elevación de concentraciones séricas de factores como TFG-alfa o de hormona de crecimiento humana en algunos pacientes. Estos factores estimularían el crecimiento epidérmico5,7,10,12. Por otro lado, Ellis et al16 demostraron, en un paciente con síndrome de Leser-Trélat asociado a melanoma cutáneo, un aumento difuso de los receptores del factor de crecimiento epidérmico (EGF) en todas las capas del estrato mucoso de Malphigio mientras que dicho receptor se localiza en condiciones normales exclusivamente en los queratinocitos basales, así como un descenso en el número de receptores, coincidiendo con la extirpación del tumor. Estas teorías explicarían la relativa frecuencia, de hasta un 35 % de los casos, con la que coexisten el signo de Leser-Trélat y la acantosis nigricans ya que, en ambos casos, la base histopatológica es igualmente una hiperplasia epidérmica7-9.
El pronóstico de los pacientes que presentan signo de Leser-Trélat depende habitualmente del tipo de neoplasia asociada. El síndrome de Sézary constituye una enfermedad de pronóstico grave y supervivencia media de 5 años. En nuestro caso, a pesar de una evolución favorable al inicio del tratamiento, la supervivencia fue de un año, probablemente debido al rechazo del paciente a continuar la terapia a los 3 meses de su inicio.
En conclusión, a pesar de su baja incidencia, creemos que la existencia de este signo paraneoplásico, es real17 y en la práctica constituye un instrumento de gran utilidad, ya que alerta al clínico de la posibilidad de una neoplasia subyacente.


