




La micosis fungoide es un linfoma de células T periféricas que se caracteriza por la infiltración y acumulación de células T tumorales en la piel1. Es el tipo más frecuente de linfoma cutáneo de células T (LCCT), y representa más de la mitad de todos los linfomas de origen en la piel2.
El diagnóstico clinicopatológico de la micosis fungoide resulta muy difícil debido al hecho de que guarda muchas semejanzas con algunas dermatosis inflamatorias que se caracterizan por infiltración y acumulación de células T benignas en la piel. Además, sobre todo en fases iniciales, el bajo porcentaje de células tumorales en la piel (muy frecuentemente por debajo del 5 % de la celularidad total) también dificulta el diagnóstico.
La mayoría de casos de LCCT tienen el fenotipo de linfocitos Th de memoria (CD3+, CD4+) y una minoría de casos tiene fenotipos como CD4 o CD8+. Los casos CD8+ posiblemente representan un subgrupo agresivo de la enfermedad3. Además, la pérdida de expresión de CD7 está considerada una característica distintiva de micosis fungoide4.
La etiología de la micosis fungoide es fundamentalmente desconocida, aunque se han propuesto algunos factores provocadores, como estimulación crónica con antígenos5 o infección con el virus de la leucemia humana (HTLV-I) u otras infecciones virales. Sin embargo, los resultados de dichos estudios han sido algo polémicos6,7. Estudios moleculares de micosis fungoide han revelado algunos resultados interesantes, aunque la mayoría de estos estudios moleculares están limitados a grupos pequeños de genes y proteínas. Se ha propuesto que la disrupción de señalización por FAS puede ser un mecanismo de la patogénesis de micosis fungoide, debido a defectos en señalización de apoptosis en células T de la piel8. Se ha descrito reducción de expresión de FAS en linfocitos CD4+ de sangre periférica en casos de LCCT9.
La reducción en la expresión de FAS puede ser el resultado de mutaciones de FAS, que no son frecuentes, pero se han descrito en algunos casos8. Los defectos en la señalización por FAS pueden ser complementados por mutaciones o defectos en la actividad de caspasas o miembros de la familia de BCL2. No obstante, estudios de BCL210 y BAX8 indican que alteraciones en estos genes no tienen un papel importante en la patogénesis de la micosis fungoide. De modo parecido, el análisis molecular de P53, el gen con más frecuencia mutado en neoplasias humanas, ha ilustrado que mutaciones en este gen están presentes únicamente en casos avanzados y es poco probable que desempeñen un papel importante en la patogénesis temprana de micosis fungoide8. Estudios sobre p16 han revelado una reducción de la expresión de p16 en lesiones de micosis fungoide progresivas a fase tumoral11,12, y algunos estudios han descrito un efecto silenciador sobre la expresión del gen p15 asociado con micosis fungoide y el síndrome de Sézary13. Estudios adicionales han implicado STAT114, STAT315, CD3016, CD40/CD40L17, IL1518, IL1618, receptores de quimocinas19 y reducción del tamaño de telómeros y actividad incrementada de telomerasas20 en la patogénesis y progresión de micosis fungoide. Últimamente, estudios in vitro han ilustrado que NF-kappa B puede ser importante en la patogénesis de micosis fungoide21.
En este estudio se han enfocado los mecanismos de tumorigénesis de micosis fungoide empleando análisis con micromatrices (chips o microchips) de ADN-c. Los datos obtenidos permiten la identificación de una firma de micosis fungoide que tiene la capacidad de distinguir entre casos de micosis fungoide y casos de dermatosis inflamatorias y es válido en un grupo aislado de casos. La técnica de micromatrices de ADN-c es una técnica muy potente que permite el análisis de la expresión de miles de genes simultáneamente. Estos estudios han revolucionado la investigación del cáncer y permiten una clasificación más precisa de los tumores22, predicción de la respuesta al tratamiento23,24 y predicción de supervivencia25.
Estos tipos de análisis se han empleado previamente en el estudio in vitro de células de micosis fungoide para identificar genes que posiblemente desempeñan un papel en el desarrollo de resistencia a interferón alfa24. Aquí, la técnica de micromatrices de ADN-c se ha utilizado para examinar el perfil de expresión génica en un total de 53 casos de micosis fungoide (29 pacientes en el grupo de estudio y un grupo de 24 pacientes adicionales para validación) y 11 casos de dermatosis inflamatorias. Los resultados permiten la identificación de 27 genes implicados en la tumorigénesis. Muchos de los genes identificados están directamente implicados en la señalización antiapoptótica por la ruta del factor de necrosis tumoral (TNF). Posteriormente, se ha identificado y validado un modelo predictivo de sólo 6 genes en el grupo de estudio (29 pacientes) y en el grupo de validación (24 pacientes).
MATERIAL Y MÉTODOS
Selección de casos
Se obtuvieron 11 muestras de dermatosis inflamatorias, 29 muestras de micosis fungoide, seis muestras de control de piel normal (no fotoexpuesta) y 24 muestras de micosis fungoide para validación en varios hospitales españoles. Todas las muestras fueron recogidas y manipuladas siguiendo un protocolo estandarizado con consentimiento informado de todos los pacientes en el estudio, supervisado por los comités éticos de los hospitales. Las muestras de micosis fungoide representan biopsias consecutivas, elegidas al azar de los hospitales colaboradores. Se revisaron centralmente por un panel de patólogos. Los diagnósticos se realizaron empleando criterios uniformes, aceptados y previamente descritos26 basados en las características clínicas, histológicas, inmunofenotípicas y moleculares, con la ayuda de datos de hematoxilina-eosina, inmunohistoquímica con CD3, CD4 y CD8, y análisis mediante reacción en cadena de la polimerasa (PCR) de reordenamiento del gen del receptor de superficie de las células T (TCR). Las muestras de micosis fungoide representan muestras iniciales de diagnóstico de pacientes con varios estadios de enfermedad incluyendo pacientes con lesiones de mancha, placa y tumor. Los pacientes con micosis fungoide del grupo de validación (24 casos) están incluidos en un ensayo clínico y representan únicamente los estadios Ia, Ib y IIa.
Las 11 muestras de dermatosis inflamatorias representan casos de dermatitis de la interfase y dermatitis espongiótica incluidos pitiriasis rosada, lupus eritematoso sistémico, dermatitis herpetiforme, dermatitis seborreica y dermatitis espongiótica.
Preparación de muestras para estudios de micromatrices de ADN-c
Se extrajo ARN total usando Trizol (Life technologies, Inc., Grand island, NY) purificado usando el método de RNeasy (Qiagen Inc., Valencia, CA) y tratado con DNase 1 (libre de ARNsas) según las instrucciones de fabricante. Se amplificaron 1-3 μg de ARN según un protocolo previamente descrito24, empleando un sistema de transcripción in vitro usando T7. Se marcaron directamente con Cy5 (cyanine 5-conjugated dUTP) 5 μg de ARN de cada muestra y 5 μg de ARN de un ARN de referencia (Universal Human Reference RNA, Stratagene, La Jolla, CA) con Cy3 (cyanine 3-conjugated dUTP) como referencia. Todos los análisis de micromatrices de ADN-c emplearon un chip de micromatrices construido en nuestro laboratorio según un protocolo previamente descrito24. La captura de imágenes se ha realizó con el Scanarray 5000 XL (GSI Lumonics, Kanata, Ontario, Canadá) y las imágenes se analizaron con el programa GenePix 4.0 Pro (Axon Instruments Inc., Union City, CA). Cada una de las muestras se analizó por duplicado. Se descartaron los resultados inconsistentes en cada una de las dos muestras analizadas.
Análisis y normalización de datos
Los datos de la intensidad de fluorescencia han sido sujetos a una resta automática de la fluorescencia de fondo y las ratios de Cy3/Cy5 se normalizaron frente a un valor mediana de la ratio de todos los puntos de la micromatriz. Se calculó la suma de las medianas del fondo y se descartaron aquellos puntos en los que el valor de intensidad total era menor que la suma de las medianas del fondo. Toda ratio se convirtió en escala LOG (base 2). También se descartaron los duplicados inconsistentes. Se calculó la media para todos los duplicados de clon y gen consistentes. Se excluyeron de análisis posteriores aquellos genes en los que se obtuvieron datos consistentes en menos de 80 % de las muestras analizadas de pacientes27.
Para reducir el fondo de señal debido a la presencia de células normales en el tejido usado, hemos utilizado dos métodos distintos: normalización por centralización de la mediana de todos los genes en cada caso de micosis fungoide y dermatosis inflamatorias y normalización de estos casos frente al grupo de muestras de piel normal no fotoexpuesta. Dado que el segundo método tuvo más éxito en cuanto a separación de los casos empleando la técnica de agrupación jerárquica, cada muestra en el estudio se normalizó frente a una colección de muestras de piel normal. Se seleccionaron seis muestras de piel normal (no fotoexpuesta) y se calculó una media para cada gen en el que estaban disponibles datos de un mínimo de cuatro muestras. Esta media se utilizó en la normalización de todos los casos de micosis fungoide y dermatosis inflamatorias.
Agrupación jerárquica y análisis estadístico
Se empleó el programa SOTA28 para los estudios de agrupación jerárquica. Los perfiles de expresión se han visualizado usando el programa TreeView. La función biológica de los genes se ha asignado usando la base de datos GENECARDS29.
Para identificar genes importantes en la distinción entre casos de micosis fungoide y casos de dermatosis inflamatorias utilizamos un valor p (la versión de Welsh que no requiere varianzas iguales) y se obtuvieron valores de p no ajustados por permutaciones. Los valores de p ajustado se obtuvieron usando el método de control de la frecuencia de falsos positivos de Benjamini y Hochberg30. Los genes con un valor de p no ajustado inferior a 0,001 y valor de p ajustado inferior a 0,1 se han considerado como genes significativos en la distinción de muestras de micosis fungoide y dermatosis inflamatorias.
Para validar el modelo predictivo de micosis fungoide, se utilizó el programa SOM31 (Self-Organizing Map Algorithm) para agrupar los 27 genes que se encontraron en los experimentos previos, definiendo un máximo de 7 grupos de genes.
La media calculada para cada grupo de genes se ha utilizado para validación mediante análisis discriminante (SPSS Base versión 10.0 [SPSS Inc., Chicago, IL]) en el grupo de pacientes del estudio (29 casos) y en el grupo de validación (24 casos). Para identificar un subgrupo reducido de genes predictivo, se seleccionó el gen con el valor de p más bajo de cada agrupación de genes identificado por SOM. El modelo predictivo de 6 genes se validó mediante análisis discriminante en el grupo de pacientes del estudio (29 casos) y en el grupo de validación (24 casos).
RESULTADOS
Normalización de datos
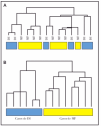
Hemos realizado estudios preliminares con 6 casos de dermatosis inflamatorias y 8 casos de micosis fungoide. El estudio de agrupamiento jerárquico no demostró separación de las muestras (fig. 1A). Teniendo en cuenta que en muchos casos de micosis fungoide y dermatosis inflamatorias el infiltrado de células T supone menos del 10 % de todas las células del tejido, tratamos de eliminar la señal debido a células normales en ambos tejidos. Para esto, intentamos dos métodos de normalización: a) normalización por centralización de la mediana de señal de todos los genes en cada caso de micosis fungoide y dermatosis inflamatorias, y b) normalización de casos de micosis fungoide y dermatosis inflamatorias frente a grupo de muestras de piel normal no fotoexpuesta. Este segundo método obtuvo resultados óptimos. En la figura 1B se puede observar cómo se separan perfectamente las señales moleculares de las micosis fungoide y las dermatosis inflamatorias (fig. 1B). Consideramos entonces que este método de normalización es válido para enriquecer los datos obtenidos de este tipo de muestras, en las que la proporción de células tumorales es baja, y permite una agrupación y clasificación de estos tumores. Durante el resto de este estudio, este tipo de normalización se ha aplicado en todos los datos de cada paciente, usando un total de seis muestras de piel normal.
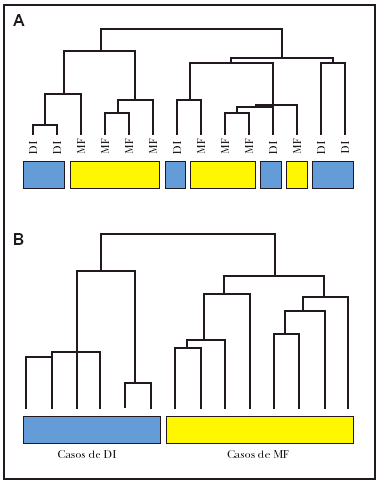
Fig. 1.--Agrupación jerárquica de datos brutos (sin normalizar) y datos normalizados de casos de micosis fungoide (MF) y dermatosis inflamatorias (DI). A) Utilizando los datos brutos (sin normalizar) en 8 casos de micosis fungoide y en 6 casos de dermatosis inflamatorias, no fue posible la separación de los grupos. B) Normalizando las muestras frente a pieles normales se consiguieron separar los casos de micosis fungoide de los de dermatosis inflamatorias.
Firma molecular de micosis fungoide: expresión diferencial con pieles inflamatorias
A fin de identificar genes implicados en la tumorigénesis de la micosis fungoide se utilizaron un total de 29 muestras de micosis fungoide y 11 muestras de dermatosis inflamatorias. Los datos de los estudios de micromatrices de ADN-c de cada u no de estos casos se normalizaron contra la señal media a partir de 6 muestras piel normal no fotoexpuesta. Usando el método de t de Student, con un valor p ajustado (tabla 1), se identificaron 27 genes significativos en la separación de casos de micosis fungoide y dermatosis inflamatorias (tabla 1).
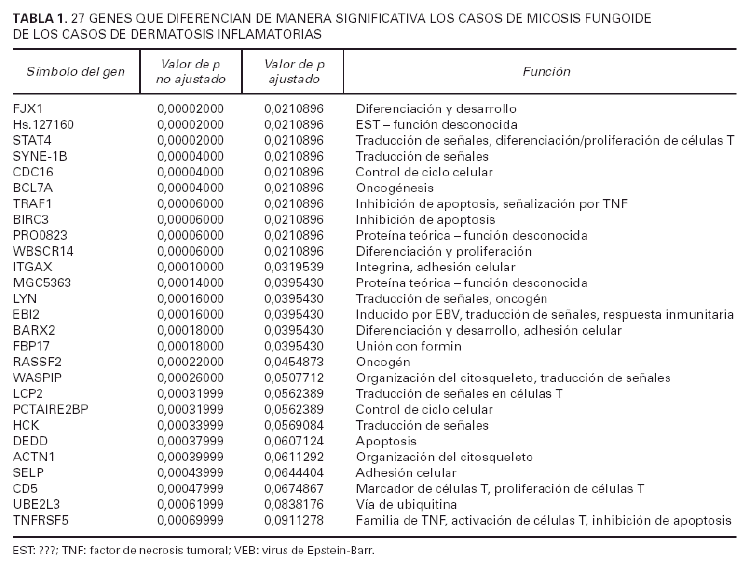
El patrón de la expresión de estos 27 genes es notablemente diferente entre los casos de ambos procesos (fig. 2). Estos genes están implicados en una variedad grande de funciones como el control del ciclo celular, apoptosis y transducción de señales. Sin embargo, el resultado más llamativo es la inducción de expresión de 7 genes implicados directamente en señalización por la vía de TNF incluyendo genes como TRAF1, BIRC3 y TNFSF5.
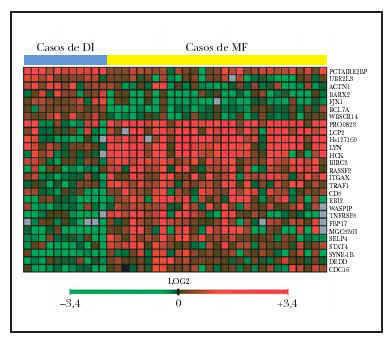
Fig. 2.--Imagen del microchip de ADN-c que muestra los 27 genes significativos en la separación de casos de micosis fungoide (MF) y casos de dermatosis inflamatorias (DI) (29 muestras de MF, 11 muestras de DI). Véase la casi perfecta separación de la imagen en cuatro cuadrantes. En rojo se representan los genes sobreexpresados. El cuadrante superior izquierdo muestra los 7 genes sobreexpresados en las muestras de DI. En el cuadrante inferior derecho, los 20 genes sobreexpresados en las muestras de MF.
Construcción y validación del modelo de predicción de micosis fungoide
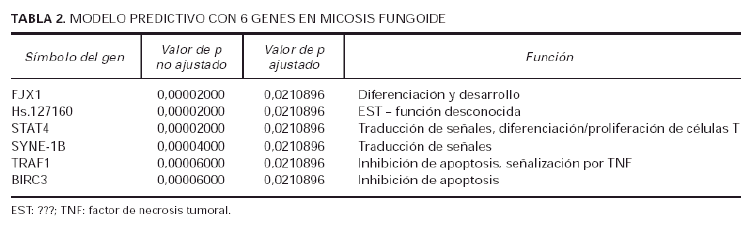
Usando el algoritmo de SOM, sobre los 27 genes significativos en la separación de casos de micosis fungoide de casos de dermatosis inflamatorias, se demostraron seis agrupamientos de genes (fig. 3), es decir, que los 27 genes pertenecían a seis familias distintas. Posteriormente, de cada una de esas familias se escogió el gen que tenía la máxima significación estadística (valor de p más bajo), con lo que se obtuvo un conjunto de 6 genes representativo del grupo original de 27 genes (tabla 2).

Fig. 3.--Agrupación de genes por el algoritmo de SOM. Selección de un modelo predictivo de micosis fungoide (MF). La agrupación de genes utilizando el algoritmo de SOM identificó 6 grupos de genes relevantes dentro del grupo de 27 genes implicados en la tumorogénesis de la MF. Para construir el modelo predictivo de MF con 6 genes se seleccionó el gen con el valor p más bajo en cada grupo.
Utilizando el grupo inicial de 27 genes, el 100 % de las muestras se podían diagnosticar perfectamente como micosis fungoide o como dermatosis inflamatorias (tabla 3). Posteriormente utilizamos este conjunto de 6 genes (tabla 2) para ver si se podía discriminar de manera aceptable entre las muestras de micosis fungoide y las de dermatosis inflamatorias. Al aplicar este pequeño grupo de genes sobre los 29 casos de micosis iniciales (que incluían lesiones en mancha, placa y tumor) y las dermatosis, el 97,3 % de los casos quedó identificado en una u otra enfermedad (tabla 3).
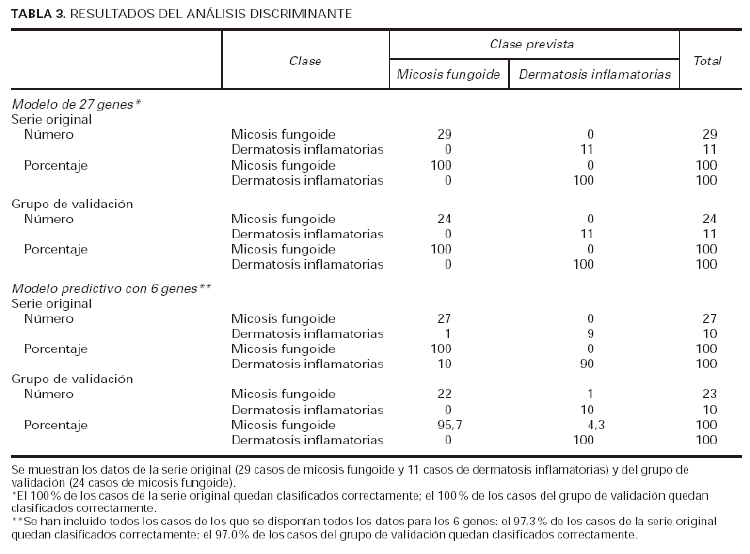
Para la validación de estos resultados, utilizamos las muestras procedentes del ensayo clínico de micosis fungoide en estadios Ia, Ib y IIa. El grupo inicial de 27 genes volvió a identificar correctamente el 100 % de las muestras (tabla 3A). Además, el grupo reducido de 6 genes permitió el diagnóstico del 97,0 % de las muestras (tabla 3B).
DISCUSION
Normalización de datos
Para entender mejor la tumorigénesis de micosis fungoide, se analizaron muestras de pacientes con esta enfermedad y las de dermatosis inflamatorias usando una micromatriz de ADN-c, que se ha diseñado específicamente para el estudio de cáncer24. Los datos en bruto de cada muestra se normalizaron a un promedio de la señal de un grupo de piel normal (no fotoexpuesta), para reducir la señal que venía de células normales en el tejido. Este paso ha conseguido reducir el ruido de fondo en estas muestras donde las células tumorales suponen menos del 10 % de las células totales y subsiguiente agrupación de muestras en subgrupos correctos (fig. 1B). Sin esta normalización, la separación de las muestras por métodos de agrupación jerárquicos no hubiera sido posible (fig. 1A).
Activación de la vía antiapoptótica del camino de TNF en la tumorigénesis de micosis fungoide
Usando los datos normalizados de micromatrices de ADN-c a partir de 29 pacientes con micosis fungoide y de 11 pacientes de dermatosis inflamatorias, se identificaron 27 genes. La expresión de los 27 genes es perceptiblemente diferente entre casos de una y otra enfermedad (tabla 1) (fig. 2). Veinte de los 27 genes identificados estaban sobreexpresados en los casos de micosis fungoide mientras que los 7 genes restantes estaban reprimidos en los casos de micosis fungoide (o lo que es lo mismo, sobreexpresados en las dermatosis inflamatorias).
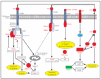
Lo más interesante es la observación de la sobreexpresión de 7 genes implicados directamente en la regulación de señalización de TNF, incluyendo BIRC3, un miembro de la familia «proteína inhibidora de apoptosis» (IAP), regulada trascripcionalmente por NF-κB32. Los genes de esta familia se caracterizan por la presencia de una región IAP o región BIR33, esencial para sus efectos antiapoptóticos32; BIRC3 o c-IAP2 poseen el dominio antiapoptótico BIR32, un dominio de CARD para unión a caspasas y un dominio RING para la degradación de proteínas diana34 tal como caspasas 3 y 735 (fig. 4). El BIRC1 también conocido como NAIP, que se une a la región citoplásmica de TNFR2, el receptor tipo 2 de TNF36. También fue encontrado como gen sobreexpresado en los casos de micosis fungoide (fig. 4). Sin embargo, la significación de esta sobreexpresión era justo fuera del rango considerado aquí (p = 0,00088; p-ajustado = 0,100698). El BIRC1, aunque carece de los dominios CARD o RING que se mencionan arriba, ejerce su acción antiapoptótica inhibiendo caspasas 3 y 7 a través de sus dominios BIR37 (fig. 4).
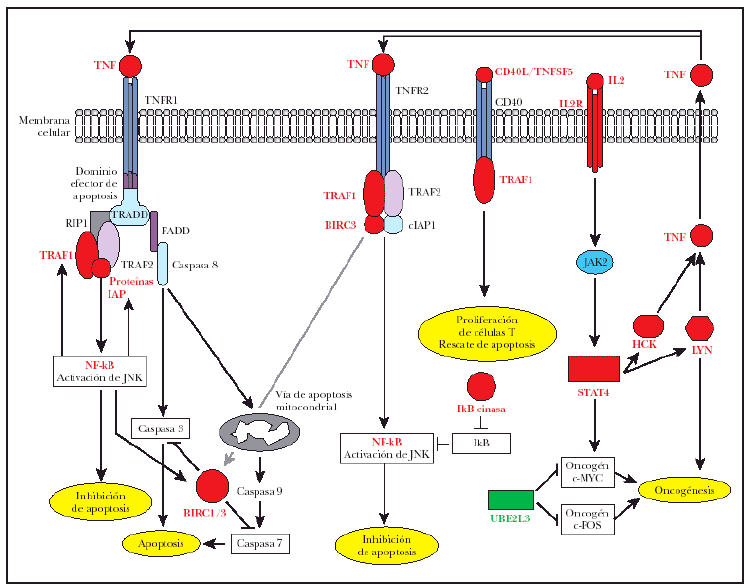
Fig. 4.--Desregulación de señalización de TNF en la tumorogénesis de micosis fungoide (MF): una combinación de señalización antiapoptótica por TNFR1, impulsado por la sobreexpresión de TRAF1 y proteínas de la familia BIRC/IAP. La señalización apoptótica de TNFR1 está reprimida por la inhibición de caspasas por BIRC1/BIRC3. La señalización antiapoptótica por TNFR2 está activada debido a la sobreexpresión de TRAF1 y BIRC3. La sobreexpresión de CD40L/TNFSF5 puede inducir proliferación de células T por el receptor de CD40 y TRAF1. La sobreexpresión de IL2R activa Jak2 y STAT4 e induce la expresión subsiguiente de los oncogenes c-MYC, LYN y HCK. Los oncogenes LYN y HCK participan en un lazo de realimentación de la señalización antiapoptótica de TNF debido a la producción endógena de TNF y la autoestimulación subsiguiente de TNFR1 y TNFR2. Texto rojo: genes sobreexpresados en casos de MF. Texto verde: genes reprimidos en casos de MF.
El TRAF1 se ha identificado aquí como un gen sobreexpresado en los casos de micosis fungoide, lo que extiende las observaciones anteriores referentes a la importancia de TRAF1 en los linfomas de células B38. El TRAF1 es inducible por NF-κB39 e interactúa con TNFR241 y varios miembros de la familia de TNF como CD4040,41 (fig. 4). TNFR2, un receptor de la superfamilia de TNF que carece del dominio intracelular de muerte celular, media la señalización de supervivencia reclutando TRAF1, TRAF236,42 y proteínas inhibidoras de apoptosis como BIRC3. El TNFR2 activa NF-κB y las vías de supervivencia de JNK36,42-44 y el complejo de proteínas de IAP/BIRC del receptor inhiben señalización apoptótica (fig. 4).
El receptor de TNF tipo 1, TNFR1, posee un dominio intracelular de muerte e induce apoptosis por el reclutamiento de TRADD y la activación subsiguiente de caspasa 8 a través de una proteína adaptadora, FADD44,45 (fig. 4). No obstante, TRAF2 se une con alta afinidad a TRADD46 y el reclutamiento subsiguiente de RIP1, TRAF1 y proteínas de la familia IAP/BIRC, conduce a la supresión de señales apoptóticas de este receptor32,46 (fig. 4).
El TNFSF5 (CD40L o TRAP) es un miembro de la familia de TNF implicado en la prevención de apoptosis y en el aumento de la proliferación celular47,48. La expresión de CD40L se ha descrito previamente en casos de micosis fungoide donde fue sugerido que CD40L pudo desempeñar un papel en un lazo paracrino importante para la prevención de apoptosis y/o regulación positiva de crecimiento17. También se ha sugerido que la expresión de CD40L puede ser importante para el acuartelamiento en piel de las células T neoplásicas17 (fig. 4).
El HCK y el oncogén LYN son tirosincinasas sobreexpresadas en casos de micosis fungoide. El HCK es responsable de la producción de TNF en células murinas49 y HCK y LYN han estado implicados en la producción de TNF en monocitos humanos50. En este estudio, además de sobreexpresión de HCK y de LYN en casos de micosis fungoide, el TNF estaba sobreexpresado, aunque la significación era justa fuera del rango riguroso usado aquí.
Además, HCK51 y LYN52 y el oncogén c-MYC53 son inducidos por IL-2 a través de la activación de JAK2 y de STAT4. En este estudio, STAT4 está sobreexpresado en los casos de micosis fungoide, al igual que el receptor IL-2 (IL2R), pero con una significación más baja. Según los resultados de este estudio, es posible que en casos de micosis fungoide, la sobreexpresión de HCK y LYN, inducido por IL2, conduzca a la producción de TNF y señalización antiapoptótica por TNFR1 y TNRF2 (fig. 4).
La UBE2L3, una proteína de la vía de ubiquitina, también conocida como UbcH7, es reprimida en esta serie de micosis fungoide. Esta proteína está implicada en ubiquitinización y degradación de los oncogenes MYC54 y c-FOS55eprimida de UBE2L3 en casos de micosis fungoide puede ayudar a la tumorigénesis de micosis fungoide (fig. 4).
Puesto que solamente los genes con un máximo de 20 % de valores desconocidos se podían incluir en este estudio, se excluyeron del análisis muchos genes con un papel posible en esta vía. Haciendo un promedio de los patrones de la expresión génica para los subgrupos de micosis fungoide y dermatosis inflamatorias para los genes con más de 20 % valores desconocidos, se proporciona una cierta evidencia de soporte para este modelo. Brevemente, NF-κB está sobreexpresado en los casos de micosis fungoide comparado con los casos de dermatosis inflamatorias (1,54 veces más expresión de NFKB1), mientras que IL2 demuestra una expresión mayor (1,86 veces más) en los casos de micosis fungoide (fig. 4). La cinasa IkB, responsable de la fosforilación de IkB y por lo tanto de la activación de NF-κB, también demostró una tendencia a estar sobreexpresado en casos de micosis fungoide. Finalmente, en estos casos también hay un aumento muy leve en la expresión de los genes implicados en las vías proliferativas y inflamatorias de JNK y p38 (JunB, JunD, MAPK14 (p38), ATF3 y TRAF2) y en las vías de la activación de NF-κB (miembros de la familia HSP90, CDC37 y TRAF2).
En conclusión, la tumorigénesis de micosis fungoide se asocia a cambios en la regulación de una combinación de señalización antiapoptótica por TNFR1 (inducido por sobreexpresi&oacut e;n de TRAF1 y BIRC/IAP) y la inhibición de las vías apoptóticas de TNFR1 (por la inhibición de la caspasa por BIRC1/BIRC3). Además, la señalización antiapoptótica por TNFR2 sigue estando activa según lo señalado por la sobreexpresión de TRAF1 y BIRC3.
La sobreexpresión de CD40L/TNFSF5 contribuye a la tumorigénesis de micosis fungoide induciendo la proliferación de células T por el receptor CD40 y TRAF1. La IL-2 también puede desempeñar un papel crítico en la tumorigénesis de micosis fungoide a través de su receptor, activando JAK2 y STAT4 y posteriormente induciendo la expresión de oncogenes c-MYC, LYN y HCK.
Finalmente, LYN y HCK participan en un lazo autocrino, participando en la producción endógena de TNF y subsiguiente activación de TNFR1 y TNFR2, lo que autoestimula la activación de la vía de supervivencia de TNF. El silencio de la vía apoptótica de TNF puede ser debido a la sobreexpresión de BIRC1/BIRC3 y la inhibición de caspasas.
Modelo predictivo de micosis fungoide
El modelo predictivo que usa la señal media de los racimos de los 27 genes puede asignar correctamente la clase (micosis fungoide o dermatosis inflamatorias) en el 100 % de la serie de micosis fungoide y en un grupo de validación de 24 casos. Posteriormente se eligió el gen con el valor p más bajo de cada racimo (fig. 3) (tabla 2) y este modelo predictivo de 6 genes clasificó correctamente 97,3 % de la serie original y 97,0 % de la serie de validación. La validación de este modelo tiene un peso adicional porque la serie de validación consta únicamente de casos de bajo estadio clínico y representan casos cuya histología se parece más a los casos de dermatosis inflamatorias, y por lo tanto, se trata de casos en los que el diagnóstico clásico es más difícil.
En conclusión, este estudio ha demostrado que, empleando estudios de micromatrices de ADN-c y normalización con muestras normales de la piel, es posible detectar una firma molecular que distingue este tipo de tumor de condiciones inflamatorias comunes de la piel. Esta firma de micosis fungoide incluye la desregulación de señalización de TNF además de un lazo autocrino de TNF que promueve señalización antiapoptótica. Además, fue posible identificar un modelo predictivo de sólo 6 genes que permite una distinción entre casos de micosis fungoide y casos de dermatosis inflamatorias en el 97,3 % de los casos en la serie original y en el 97,0 % de los casos en una serie de validación de 24 pacientes con micosis con estadios bajos.
AGRADECIMIENTOS
Gracias a Isabel Fernández y Mar López por su ayuda con la extracción del ARN del tejido congelado y de la amplificación subsiguiente. Gracias también a los miembros del departamento de análisis de microarray por la ayuda con la preparación e hibridación de las muestras. Agradecemos a Ramón Díaz y Javier Herrero, ambos de la unidad de bioinformática del CNIO, su ayuda y consejo sobre métodos estadísticos y agrupación jerárquica, respectivamente.
A este trabajo se le concedió el premio Academia Española de Dermatología 2003.
Ha sido financiado por concesiones del Ministerio de Ciencia y Tecnología (Bio2000-0275-c02/01 y/02, Saf2001-0060), España. L.T. tiene becas del Centro Nacional de Investigaciones Oncológicas (CNIO) y del Departamento de Hematología y el Instituto de Medicina Molecular, Hospital St. James, Dublín, Irlanda (beca PRTLI del Higher Education Authority de Irlanda).


