
INTRODUCCIÓN
En 1930, Brusting y cols. (1) introdujeron el término pyoderma gangrenosum, que se ha impuesto casi universalmente salvo entre algunos autores de lengua francesa que siguen denominándola piodermitis fagedénica.
La piodermia gangrenosa (PG) es una entidad que se engloba actualmente en el contexto de las dermatosis neutrofílicas; constituye una enfermedad inflamatoria, destructiva, crónica, no infecciosa y, a pesar de lo que su nombre indica, no es una piodermitis bacteriana ni una gangrena vascular. La etiopatogenia sigue siendo desconocida, pero se ha descartado la naturaleza infecciosa que inicialmente le atribuyeron Brusting y cols. (1) y últimamente la mayoría de los autores la consideran como manifestación de una función inmunitaria alterada (2).
El aspecto clínico característico de la lesión ulcerosa permite hacer el diagnóstico, ya que no existen datos analíticos ni histopatológicos específicos. Su evolución es crónica, con remisiones y exacerbaciones y con lenta resolución, dejando como lesión residual una cicatriz atrófica o cribiforme con hiper o hipopigmentación.
Aunque la PG puede aparecer sin trastornos extracutáneos asociados (3, 4), en el 40 al 60% de los casos se asocia a una enfermedad sistémica subyacente: colitis ulcerosa, enfermedad de Crohn, artritis reumatoide, leucemia mieloblástica, policitemia vera, linfomas, gammapatías monoclonales, mieloma, hepatitis crónica activa o sarcoidosis. Por tanto, el diagnóstico debe ir siempre seguido de exámenes complementarios dirigidos a la búsqueda de posibles enfermedades asociadas.
DESCRIPCIÓN DEL CASO
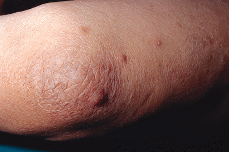
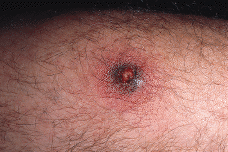
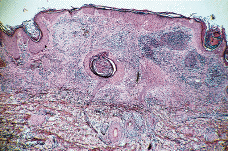
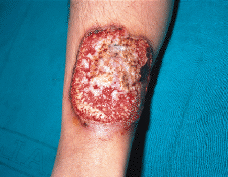
Paciente masculino de 50 años de edad con el único antecedente personal de interés de hipertrigliceridemia controlada con dieta. Consultó al Servicio de Dermatología en el año noventa por presentar lesiones papulotuberosas, algunas pustulosas, en ambos codos, de varios meses de evolución (Figs. 1 y 2). La analítica de rutina solicitada, salvo los triglicéridos, fue rigurosamente normal. El cultivo de hongos, bacterias, tinción y cultivo para micobacterias y la prueba de Mantoux resultaron negativos; la radiografía de tórax sin alteraciones. En el estudio histológico de una de las lesiones de antebrazo se observó necrosis en dermis con infiltrado abcesiforme y algunas células linfohistiocitarias (Fig. 3); no se llegó a un diagnóstico definitivo. Un mes después apareció en pierna izquierda una lesión ulcerosa de 6-7 cm de diámetro con necrosis central y borde eritematovioláceo, lo que orientó hacia el diagnóstico definitivo de PG (Fig. 4). Se inició entonces tratamiento con corticoesteroides por vía oral (prednisona, 60 mg/día) con lo que se consiguió la reepitelización progresiva de la lesión de la pierna y el aplanamiento de las lesiones de antebrazos. El estudio del aparato digestivo para descartar la presencia de enfermedad inflamatoria intestinal (EII) resultó negativo, pero se detectó la presencia de una úlcera en estómago y duodeno que obligó a suspender la corticoterapia oral; esto originó empeoramiento y la aparición de un nuevo brote de lesiones.
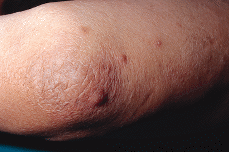
FIG.1.--Lesiones papulotuberosas y pustulosas en codo y antebrazo con las que debutó el paciente.
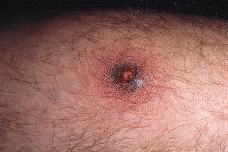
FIG. 2.--Detalle de una lesión pustulosa rodeada de halo inflamatorio eritematovioláceo.
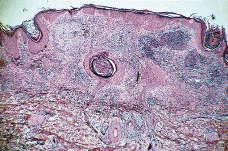
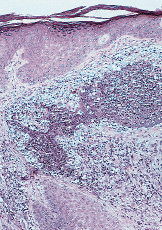
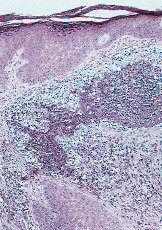
FIG. 3.--A: Estudio histológico con hematoxilinaeosina: bajo una epidermis acantósica se observa en dermis foco de necrosis con infiltrado abcesiforme de predominio de neutrófilos y algunas células linfohistiocitarias. B: Imagen a detalle del infiltrado abcesiforme en dermis.
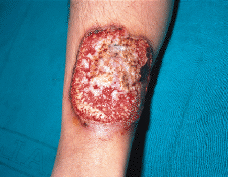
FIG. 4.--Lesión ulcerosa en pierna típica de PG con fondo granulante y borde sobreelevado violáceo.
Se inició tratamiento con sulfasalazina (4 g/día) mejorando claramente las lesiones y permitiendo disminuir gradualmente la dosis hasta alcanzar una dosis mínima de mantenimiento de 1,5 g/día, por debajo de la cual aparecía un nuevo brote que obligaba a aumentar la medicación.
En febrero de 1995 el paciente presentó en cara posterior de pierna izquierda una nueva lesión ulcerosa típica de PG que alcanzó el tamaño de 10-12 cm de diámetro y de acuerdo con el paciente se inició tratamiento con ciclosporina a dosis de 6 mg/kg/día.
En estas fechas se detectaron valores de inmunoglobulina Ig A de 600-700 mg% que, valorados por el Servicio de Hematología, llevaron al diagnóstico de gammapatía monoclonal IgA de significado incierto de cadena ligera Kappa, sin infiltración de médula ósea, proteinuria de Bence-Jones negativa y con serie ósea normal, sin ninguna repercusión clínica.
A los 2 meses de iniciado el tratamiento, y estando con esas dosis de ciclosporina, apareció un nuevo brote de lesiones ulcerosas en codos, ingle, región clavicular y pierna que obligaron a asociar 6 g/día de sulfasalazina y a la disminución progresiva de la ciclosporina.
En los 4 últimos años el paciente se ha mantenido bien controlado con 1,5-2 g/día de sulfasalazina, sin lesiones ulcerosas, y con la aparición de alguna lesión papulosa en extremidades que se ha controlado aumentando la medicación según precisaba.
Los estudios digestivos de control insistiendo en la búsqueda de EII dada la buena respuesta a la sulfasalazina siguen siendo negativos y la paraproteinemia, controlada por el Servicio de Hematología, permanece estable, sin transformación maligna tras 5 años de su diagnóstico.
DISCUSIÓN
El diagnóstico de PG es eminentemente clínico. Se trata de una lesión ulcerosa con fondo granulante y necrótico, con pequeños abcesos estériles, borde sobreelevado violáceo cortado a pico por el lado interno y en pendiente suave hacia fuera, rodeado en la periferia de una zona inflamatoria eritematosa mal definida. Antes de alcanzar este estadio característico puede iniciarse como nódulo violáceo o papulopústulas de aspecto forunculoide como en el caso que aquí se describe.
La lesión es habitualmente única, pero pueden ser múltiples y confluentes. Las localizaciones predominantes son nalgas, extremidades inferiores y cara. Pueden ser muy dolorosas o indoloras y acompañarse o no de fiebre y síntomas generales (5). Una característica importante es la aparición de nuevas lesiones ante traumatismos mínimos, muy a menudo sobre prominencias óseas o intervenciones quirúrgicas (fenómeno de patergia), aunque, según algunos autores, ésto se observa sólo en un 20% de los pacientes (6). Las formas postquirúrgicas de PG se han descrito en pacientes con PG ya conocida o con enfermedades clásicamente asociadas a PG y también, aunque es menos frecuente, en pacientes sin ningún antecedente previo de enfermedad cutánea o extracutánea asociada, denominándose en este caso piodermia gangrenosa postquirúrgica idiopática (7).
En la PG no existen datos de laboratorio específicos y la histología también es inespecífica. La célula fundamental en el infiltrado inflamatorio es el neutrófilo. En general la epidermis y la dermis superficial suelen estar ulceradas y necróticas y existe un denso infiltrado abcesiforme de neutrófilos que suele extenderse hasta hipodermis; la epidermis del borde de la úlcera se muestra hiperplásica, lo que es más evidente en lesiones evolucionadas y el infiltrado será tanto más crónico, linfocitario, cuanto más nos alejemos del borde activo de la lesión. Su y cols. (8) reconocen tres patrones histológicos dependiendo del lugar de la biopsia.
El diagnóstico de PG exige hacer previamente el diagnóstico diferencial mediante los cultivos microbiológicos y estudios histológicos correspondientes, con la gangrena postoperatoria, infecciones por clostridios, micobacterias, micosis profundas, úlceras tropicales, pénfigo vegetante y úlceras de estasis.
En 1988, Wilson-Jones y Wilkeman describieron por vez primera una variante clinicopatológica de la PG clásica que denominaron piodermia granulomatosa superficial (PGS). Esta entidad aún poco conocida tiene una serie de características clinicoevolutivas, rasgos histológicos, localización y respuesta terapéutica, que la diferencian de la PG clásica (9). Si consideramos la piodermia gangenosa como un espectro clínico, la PG clásica se situaría en un extremo y la PGS, benigna e indolente, en el extremo opuesto.
La alteración humoral más frecuentemente descrita en la PG es la existencia de gammapatía monoclonal de significado incierto, del tipo IgA. El mieloma múltiple, también generalmente IgA, puede presentarse en estos pacientes (10).
La asociación de PG y gammapatía monoclonal es bien reconocida, con una incidencia de alrededor del 10% (11), pero la relación patogénica de la paraproteinemia y las lesiones cutáneas sigue siendo una incógnita. En estudios de series largas de pacientes con gammapatía monoclonal como la aportada por Kyle de 241 pacientes, el 75% fueron IgG, 15% IgM y el 10% IgA (12); en cambio la gammapatía monoclonal IgA es el tipo más frecuentemente asociado a PG (13, 14). La gammapatía monoclonal es una enfermedad cuya incidencia aumenta con la edad y por tanto la PG asociada a ésta se presenta en edades más avanzadas que en la forma idiopática (13, 14). La secuencia de aparición de la PG y de la gammapatía es variable. En nuestro paciente, como en otros recogidos en la literatura, las lesiones de PG preceden al diagnóstico de la gammapatía, pero hay otros muchos casos en que la secuencia de aparición es inversa o simultánea (15).
La paraproteinemia IgA se asocia además con frecuencia a otras dermatosis neutrofílicas (16-18), como la pustulosis subcórnea y el erythema elevatum et diutinum.
Se sabe que los leucocitos neutrófilos tienen receptores de superficie para la IgA, la cual unida en su superficie inhibe la actividad quimiotáctica de los neutrófilos y favorece la acumulación local en los tejidos. Van Epps y cols. (19) demostraron cómo las inmunoglobulinas IgA inhiben la función de los neutrófilos in vitro; Holt y y cols. (20) encontraron reducida la quimiotaxis leucocitaria, el retraso en la migración leucocitaria y una anormal capacidad bactericida; Lazarus y cols. (21) encontraron fallos en la hipersensibilidad retardada en pacientes con PG y gammapatía monoclonal.
Todo esto hace pensar en la existencia de un vínculo etiopatogénico entre la disfunción IgA y las dermatosis neutrofílicas, pero hasta el momento sigue siendo una hipótesis donde quedan muchos puntos por aclarar.
Es importante el seguimiento periódico de los pacientes con PG y «gammapatía monoclonal de significado incierto» puesto que no existen medios fiables para conocer si esta gammapatía permanecerá estable o evolucionará a mieloma, hecho poco frecuente pero posible (11).
La PG ha recibido múltiples tratamientos tanto locales como sistémicos con resultados inconstantes. Como tratamientos locales se han utilizado povidona yodada, nitrato de plata, permanganato potásico y corticoesteroides intralesionales, y como terapeútica sistémica, corticoesteroides orales (para la mayoría el tratamiento de elección), dapsona, sulfasalazina, clofazimina y minociclina. En los últimos años se ha demostrado la eficacia de los inmunosupresores en el tratamiento de la PG y la ciclosporina ha sido sin duda el más eficaz. El primer caso de PG con respuesta favorable a la ciclosporina fue descrito por Curley y cols. en 1985 (22) y desde entonces múltiples publicaciones han corroborado su eficacia (4, 23-26).
La sulfasalzina es la terapéutica a la que mejor ha respondido nuestro paciente, aunque algunos trabajos comentan que la buena respuesta se da cuando la PG se asocia a colitis ulcerosa; en otros, como también ocurrió en nuestra aportación, el tratamiento ha sido eficaz sin existir tal asociación (27). Las dosis iniciales son de 4 - 6 g/día y tras la respuesta terapéutica se reduce gradualmente a una dosis de mantenimiento de 0,5-1 g/día (28). La sulfasalazina está compuesta por una sulfamida (sulfapiridina) que se comporta como molécula transportadora unida por un enlace azoico al ácido 5- aminosalicílico (5-ASA) que es el producto activo. La acción del 5-ASA es inhibir la función de las prostaglandinas, afectando también a la actividad de los neutrófilos a través de los inhibidores de la lipooxigenasa cuyos metabolitos, los leucotrienos, tienen una importante función quimiotáctica sobre los neutrófilos.
Aportamos, por tanto, un nuevo caso de PG asociada a gammapatía monoclonal IgA benigna o de significado incierto que además presentaba las siguientes peculiaridades que nos parecen interesantes: a) el paciente debutó inicialmente con lesiones papulo-tuberosas y pustulosas que demoraron en 1 mes el diagnóstico correcto, hasta la aparición de la lesión ulcerosa típica de PG en la pierna; b) el diagnóstico de PG precedió al diagnóstico de la gammapatía monoclonal benigna IgA; c) evolución favorable de la gammapatía sin transformación a mieloma tras 5 años de seguimiento; d) buena respuesta a la sulfasalazina sin tener asociado enfermedad inflamatoria intestinal, y e) el paciente presentó a los 40 años lesiones de PG y a los 45 años se diagnosticó la gammapatía monoclonal, edad más temprana que en otros casos aportados en la literatura con esta asociación.


