

INTRODUCCION
La histiocitosis de células de Langerhans (HCL) es una enfermedad poco frecuente que incluye un grupo polimorfo de alteraciones clínicas caracterizadas por la proliferación clonal de células con el fenotipo de las células de Langerhans (1). Estas células pueden infiltrar virtualmente cualquier lugar del organismo, produciendo una lesión localizada o una enfermedad sistémica (2).
La HCL puede ocurrir a cualquier edad, pero su inicio es más común entre 1 y 15 años. La incidencia en la población anciana es excepcional. En estos casos el diagnóstico clínico es más difícil, pero generalmente el pronóstico es mejor (2-10).
La HCL es una enfermedad de etiología desconocida (11); existe gran controversia respecto a si se trata de un proceso reactivo o neoplásico. Actualmente la opinión mayoritaria es la de considerarla como un proceso neoplásico clonal que procede de mutaciones de células precursoras de la médula ósea (12-14).
Se ha descrito la asociación de HCL y leucemias de la línea mielomonocítica (15-17). Como los sistemas monocito/macrofágico y las células de Langerhans tienen un origen común, una alteración de la célula madre pluripotencial podría explicar la coexistencia de ambos procesos (15).
Presentamos un caso de HCL en un paciente anciano, con afectación cutánea extensa y ganglionar, que posteriormente se asoció a una leucemia de la línea mielomonocítica (leucemia mielomonocítica crónica), sin existir previamente ningún tratamiento capaz de inducirla.
DESCRIPCIÓN DEL CASO
Varón de 76 años con antecedentes personales de diabetes mellitus no insulindependiente e infarto agudo de miocardio que estaba en tratamiento con antidiabéticos orales y ácido acetilsalicílico. Acudió a nuestras consultas en junio de 1999 por unas lesiones cutáneas de 5 meses de evolución, levemente pruriginosas, sin sintomatología general asociada. Tres meses antes le habían extirpado un pólipo del conducto auditivo externo derecho, cuyo diagnóstico histopatológico fue sugerente de HCL, sin que ello se relacionara entonces con las lesiones cutáneas que ya presentaba, ni se le implantara ningún tratamiento sistémico.
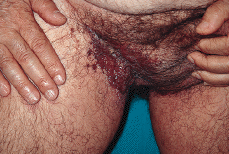
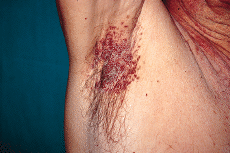
En la exploración física se observaron lesiones cutáneas de distribución generalizada, de predominio en regiones seborreicas y en grandes pliegues (Figs. 1 y 2), sobre todo en ingles, en forma de placas infiltradas, de coloración violácea y superficie exudativa y erosionada. Presentaba también lesiones papulosas, de coloración rojiza-marrón, algunas amarillentas, con tendencia a confluir, localizadas a nivel de región anterior del tronco, cuello, región abdominal y surcos retroauriculares. También existían lesiones purpúricas en mucosa oral y descamación untuosa blancoamarillenta en cuero cabelludo. En cuello, axilas e ingles se palpaban múltiples adenopatías.
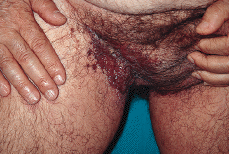
FIG. 1.--Placa infiltrada con superficie exudativa y erosionada, de coloración violácea, en región inguinal derecha.
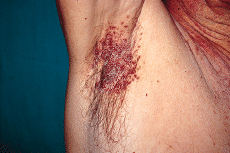
FIG. 2.--Pápulas confluentes en axila derecha.
Se realizaron biopsias cutáneas que mostraban infiltración densa, masiva, por células con citoplasmas claros y núcleos irregulares, arriñonados (Fig. 3), localizadas tanto a nivel profundo como superficial, pegado a la epidermis, donde penetraban formando tecas o como células aisladas. Estas células se mezclaban con un infiltrado inflamatorio compuesto por linfocitos, células plasmáticas y escasos eosinófilos. Se realizó estudio inmunohistoquímico que demostró positividad para CD43, CD68, CD1a y S100. El estudio ultraestructural fue compatible con HCL, con frecuentes elementos sugerentes de ser células de Langerhans, pero sin evidenciar gránulos de Birbeck.
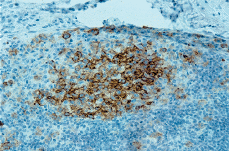
FIG. 3.--Infiltración difusa, en sábana, de células con citoplasmas claros y núcleos irregulares, arriñonados, que ocupa la dermis superficial y profunda junto con un infiltrado inflamatorio polimorfo (hematoxilina-eosina).
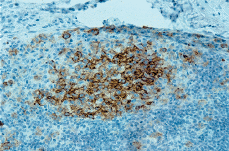
Posteriormente se realizó biopsia de adenopatía inguinal que demostró infiltración por una HCL. El estudio histológico mostraba la presencia de una proliferación de células histiocitarias de aspecto epitelioide, con núcleos de contornos irregulares, ocasionalmente vacuolados, con fisuras intranucleares y nucléolos evidentes; entre ellas se distinguían abundantes cuerpos apoptóticos. En el estudio inmunohistoquímico del ganglio se evidenció positividad para CD1a (Fig. 4) y S100 en las células del infiltrado, fenotipo compatible con HCL.

FIG. 4.--Las células que infiltran el ganglio muestran positividad para el CD1.
Con el diagnóstico de HCL cutánea y ganglionar se realizó estudio de extensión para descartar afectación multisistémica. En las pruebas complementarias realizadas destacaba en el hemograma una leucocitosis de 13.610 leucocitos/µl (normal: 4.000-10.300), con 2.670 monocitos/µl (normal: 200-1.000); el inmunofenotipo en sangre periférica mostraba una línea monocítica aumentada, con un valor de 2.350/mm3 [CD14+, CD33+, CD15+ (15%), CD11C+] y presencia de promonocitos (12%). El estudio de médula ósea descartaba la invasión por células de Langerhans (CD1a y S100 negativas), siendo compatible con un síndrome dismielopoyético tipo leucemia mielomonocítica crónica. El estudio citogenético de la médula ósea mostraba una línea celular con deleción parcial o translocación del brazo corto del cromosoma 7 en el 13% de las células analizadas −46,XY del/t (7p)−, y una línea celular normal en el 87% de las células −46,XY−. El resto de pruebas complementarias realizadas (bioquímica, sistemático de orina, velocidad de sedimentación, espectro electroforético, radiografía de tórax, electrocardiograma, serie ósea, gammagrafía, TAC craneal y toracoabdominal) fueron normales o presentaban mínimas alteraciones no significativas.
DISCUSIÓN
La HCL es una entidad poco frecuente que se presenta generalmente en niños, siendo muy rara en adultos (3-7), donde es difícil de diagnosticar clínicamente.
Stefanato y cols. (8) presentaron tres casos de HCL en ancianos y realizaron una revisión desde 1966 hasta 1996, encontrando 55 casos de HCL en la edad adulta de un total de 1.350 casos de HCL.
La etiopatogenia de esta enfermedad continúa siendo un misterio pese a los numerosos estudios de investigación realizados (11). Se han intentado implicar factores virales (18), inmunológicos (19-21) y neoplásicos, sin que ninguno de ellos pueda considerarse como indudable factor etiológico. Su carácter neoplásico o reactivo está ampliamente discutido en la actualidad.
El análisis de DNA por citometría de flujo (22-24) no ha podido identificar, excepto en un caso (25), aneuploidía en la HCL. Recientes estudios (12, 13) han demostrado la clonalidad de las células CD1a+ del infiltrado en las distintas formas clínicas de la HCL, lo que proporciona una fuerte evidencia de la naturaleza neoplásica, aunque no necesariamente maligna de esta enfermedad.
La HCL está raramente asociada a neoplasias hematopoyéticas malignas, pero se ha descrito la asociación con leucemia aguda o linfoma (2). Egeler y cols. (15) hicieron una revisión de la literatura de la asociación de HCL con neoplasias malignas, encontrando 22 casos de HCL asociados a leucemias; de éstos, el 77% eran de la línea mielomonocítica y sólo dos de ellos mayores de 60 años.
La posibilidad de generar segundas neoplasias con los distintos agentes quimioterápicos utilizados para el tratamiento del cáncer está ampliamente demostrada (26).
Baikian y cols. (17) presentaron en 1999 tres casos de HCL asociadas a leucemias de la línea mielomonocítica, similares a nuestro paciente, en los que no existían quimioterápicos sistémicos que potencialmente pudieran producir leucemias; esta asociación sugiere para ellos un origen común para ambos procesos por alteración de la célula madre pluripotencial. Se trataba de pacientes de 71, 73 y 75 años con HCL que también desarrollaron leucemias de la línea mielomonocítica (LAM4, LMMC y LAM5, respectivamente). En el segundo (muy semejante al nuestro) y tercer casos no se habían realizado tratamientos previos capaces de inducir leucemia. En el primer caso, la LAM4 se desarrolló poco tiempo después del comienzo de tratamiento con vinblastina, pero el período de latencia era demasiado corto para considerar que la leucemia era secundaria al quimioterápico. Para Baikian y cols. esta asociación y la existencia de casos familiares, en particular dos gemelos monocigotos (27), refuerzan la hipótesis de que la HCL es más una afectación de origen genético que reaccional.
Los sistemas monocito/macrofágico y las células de Langerhans/células dendríticas presentan algunos antígenos celulares comunes (CD1a, CD11, CD45). Ambos sistemas tienen un origen común, derivan de células madre medulares CD34+ del sistema fagocítico mononuclear (28); por tanto, las células de la HCL pueden también originarse de los mismos progenitores hematopoyéticos pluripotenciales que las leucemias de la línea mielomonocítica. Una alteración en la célula madre pluripotencial de la línea del sistema fagocítico mononuclear podría ser responsable de ambas entidades una vez excluidas neoplasias secundarias a quimioterápicos.
En base a estos hallazgos la opinión mayoritaria actualmente es la de considerar a la HCL como un proceso neoplásico clonal, procedente de mutaciones somáticas de precursores de la médula ósea, que promueven la expansión clonal de células de Langerhans o sus progenitores en la médula ósea y otros órganos, con un comportamiento biológico altamente variable. Estas mutaciones genéticas todavía no han sido identificadas. Los excepcionales casos familiares hacen improbable que una mutación predisponente pudiera ser heredada como en otras neoplasias de la infancia (12).
Quizá el caso de esta asociación descrita entre HCL y leucemias de la línea mielomonocítica podría tratarse de algo similar a lo que ocurre en la neutropenia congénita severa, donde hay una mutación sin sentido del gen del receptor de G-CSF que contribuye tanto al desarrollo de esta enfermedad como al de leucemia mieloide aguda (29).
Nosotros presentamos un nuevo caso de un varón de 76 años que presenta una HCL cutánea extensa con afectación ganglionar que posteriormente se asoció a una leucemia de la línea mielomonocítica (leucemia mielomonocítica crónica) sin haber existido previamente ningún tratamiento capaz de inducirla.


