



Sr. Director:
El síndrome orofaciodigital (SOFD) tipo I, o síndrome de Papillon-Léage Psaume (MIM 605041), se caracteriza por la presencia de malformaciones orofaciales y digitales.
Es frecuente que los pacientes desarrollen alteraciones dermatológicas durante los primeros meses de vida. El papel del dermatólogo puede ser clave para establecer un diagnóstico precoz. La enfermedad aparece en un 75 % de los casos de forma esporádica, siguiendo el resto una herencia dominante ligada al cromosoma X. Es letal en varones, por lo que sólo se observa en mujeres. Su incidencia es de 1/50.000 recién nacidos vivos1. El gen responsable de la enfermedad se encuentra en el brazo corto del cromosoma X (Xp22.2-22.3). El producto genético de este fragmento podría tener un papel fundamental en la organogénesis2.
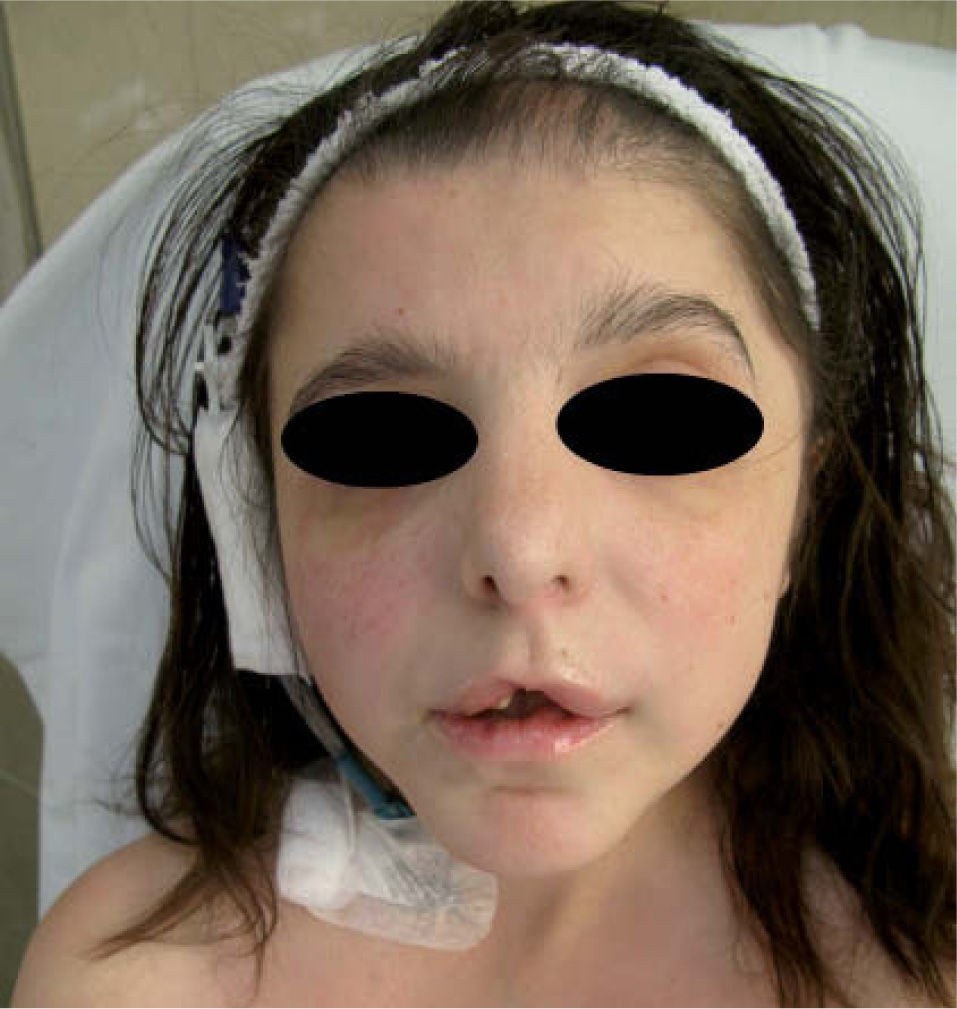
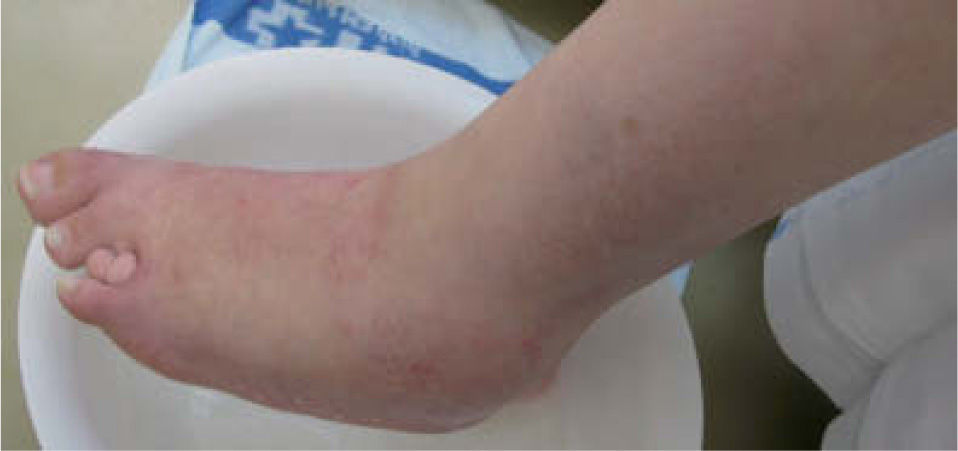
Presentamos el caso de una mujer de 29 años de edad, ingresada en el Servicio de Nefrología por rechazo vascular de un trasplante renal. Consultaba por prurito generalizado y sequedad cutánea acentuados durante el ingreso hospitalario. En la exploración física destacaban una facies anómala con hipertelorismo, una raíz nasal ancha, una implantación baja de los pabellones auriculares, una hendidura en el labio superior y la ausencia de múltiples piezas dentarias (fig. 1). Presentaba, además, braquidactilia en los dedos de ambas manos y en algunos de los de los pies (fig. 2). El pelo era fino, seco y frágil, con áreas de menor densidad capilar, más evidentes en la línea de implantación frontal.
La paciente era fruto de un embarazo sin complicaciones y un parto eutócico. No existía consanguineidad entre los progenitores y el cariotipo resultó 46XX normal. Desde el momento del nacimiento presentaba múltiples anomalías fenotípicas, destacando una lengua trilobulada con frenillo supernumerario, anomalías dentarias, una hipoplasia de los cartílagos alares, braquidactilia y áreas de alopecia difusa en el cuero cabelludo. Fue sometida a múltiples cirugías reparadoras durante su infancia. La pérdida progresiva de piezas dentarias dificultaba actividades básicas tales como la masticación o la emisión del lenguaje. A los 17 años desarrolló una insuficiencia renal por la presencia de múltiples quistes y requirió finalmente un trasplante renal. Nuestra paciente presentaba xerosis cutánea y prurito, acentuados por el deterioro de la función renal. Se recomendó tratamiento con antihistamínicos orales y el uso de emolientes.
En 1954 Papillon-Léage y Psaume describieron un síndrome con malformaciones faciales, orales y digitales3. Posteriormente, al heterogéneo grupo de síndromes que incluían malformaciones en dichas localizaciones se les designó como SOFD 4. Se han descrito 13 variantes, siendo complicada la identificación de cada una de ellas por presentar características y rasgos comunes. El síndrome descrito por Papillon-Léage y Psaume es conocido como SOFD tipo I, y supone la variante más frecuente. Sólo los tipos I y II presentan alteraciones cutáneas5. Las anomalías en la cavidad oral son las más constantes y características. Los pacientes pueden presentar una lengua lobulada, un frenillo corto o supernumerario, anomalías del paladar blando y duro, del suelo de la boca y de las encías, dientes supernumerarios y ausencia de piezas dentarias. Es característica una hendidura del labio superior en la línea media. Las anomalías faciales incluyen una frente prominente, hipertelorismo, una raíz nasal ancha, hipoplasia de los cartílagos alares y de los maxilares, micrognatia e implantación baja de los pabellones auriculares 6. Las alteraciones digitales más frecuentes son la clinobraquisindactilia y la polidactilia. La imagen radiológica de las falanges muestra una radiolucencia irregular reticulada en la parte media de las mismas, hallazgo considerado como patognomónico del SOFD tipo I 7. Un 50 % de los pacientes presenta retraso mental o alteraciones del sistema nervioso central. La afectación visceral condiciona el pronóstico. Hasta un 15 % presenta quistes renales que pueden conducir a una insuficiencia renal terminal 8. También pueden aparecer quistes hepáticos y pancreáticos.
Las alteraciones dermatológicas resultan de gran utilidad para establecer un diagnóstico precoz. La aparición de múltiples quistes de milio en el cuero cabelludo, la cara y el dorso de las manos es frecuente en los primeros años de vida. Se resuelven dejando cicatrices puntiformes en las áreas afectadas9. Más de la mitad de los pacientes presentan una alopecia difusa con pelo fino, seco y frágil. Es típico observar pelos fragmentados en la línea de implantación del pelo. Presentan, además, xerosis cutánea y anomalías en los dermatoglifos de los dedos10.
Establecer un diagnóstico precoz es fundamental ante un recién nacido con malformaciones. El hallazgo de anomalías faciales, orales y digitales asociado a alteraciones cutáneas debe hacer sospechar el diagnóstico de SOFD tipo I. Los quistes de milio sólo están presentes en esta variante. Es necesario un enfoque multidisciplinar. En los primeros años de vida deben realizarse cirugías reparadoras de las malformaciones para evitar secuelas en el desarrollo y maduración del paciente. Deben seguir controles odontológicos periódicos para evitar la pérdida de piezas dentarias. El cuidado dermatológico requiere el uso frecuente de emolientes y una alimentación equilibrada que evite un déficit de nutrientes. Asimismo, deben realizar controles periódicos de las funciones renal y hepática para detectar precozmente una enfermedad poliquística.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.


