


Varón de 28 años, sin antecedentes familiares de interés, que acude por la presencia de nódulos en las manos, asintomáticos, los cuales habían aparecido desde la adolescencia. Como antecedentes presentaba un panhipopituitarismo secundario tras la exéresis de un encondroma cerebral, y había sido intervenido de un alargamiento de fémur y tibia derechos.
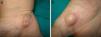
Exploración físicaPresentaba 2 tipos de nódulos subcutáneos: unos de consistencia blanda, móviles y de coloración violácea, que tenían un aspecto vascular (fig. 1a) y otros indurados al tacto, no desplazables y de coloración de la piel normal, que sugerían un origen óseo (fig. 1b). Además destacaba la presencia de una escoliosis marcada, así como dismetría, con acortamiento y deformidad en varo de las extremidades del hemicuerpo derecho.
Histopatología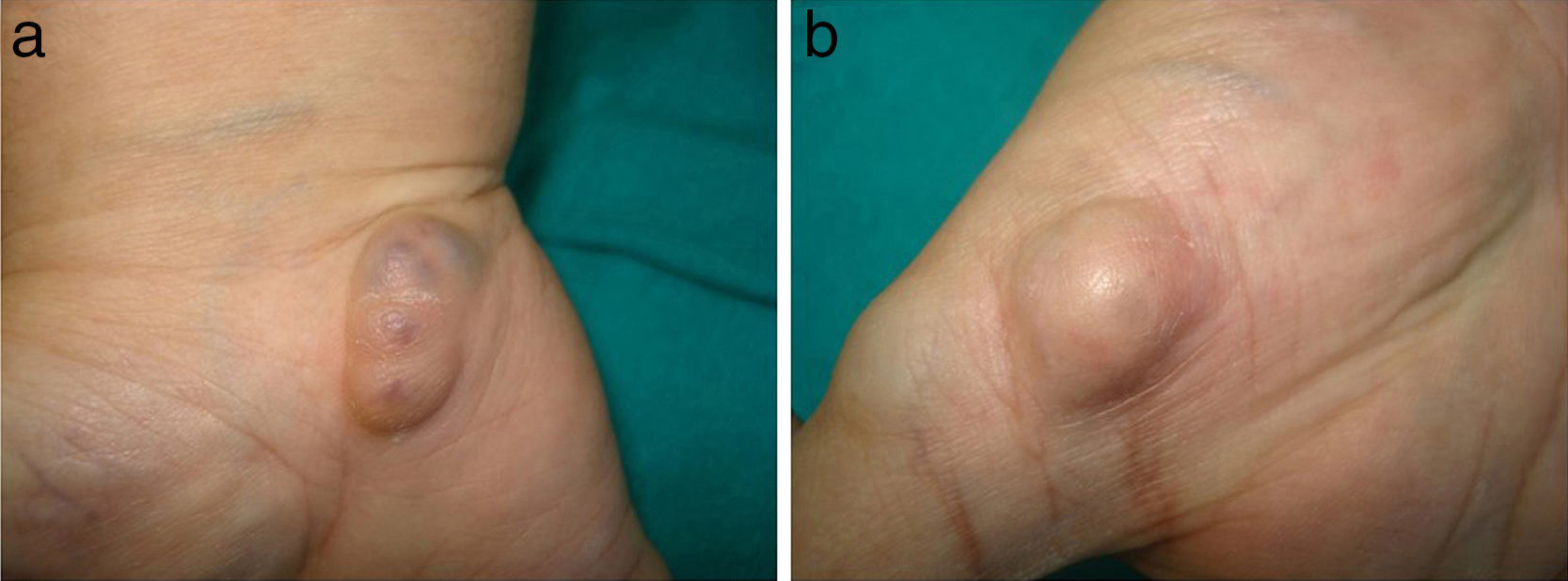
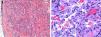
La biopsia de una lesión de aspecto vascular mostró un nódulo bien delimitado, localizado en la dermis; constituido por canales vasculares dilatados delimitados por un endotelio fino. Además se apreciaban unas áreas más sólidas, formadas por fascículos de células fusiformes (fig. 2a), entre los cuales destacaban unas células redondeadas con citoplasma vacuolado (fig. 2b).
Otras pruebas complementarias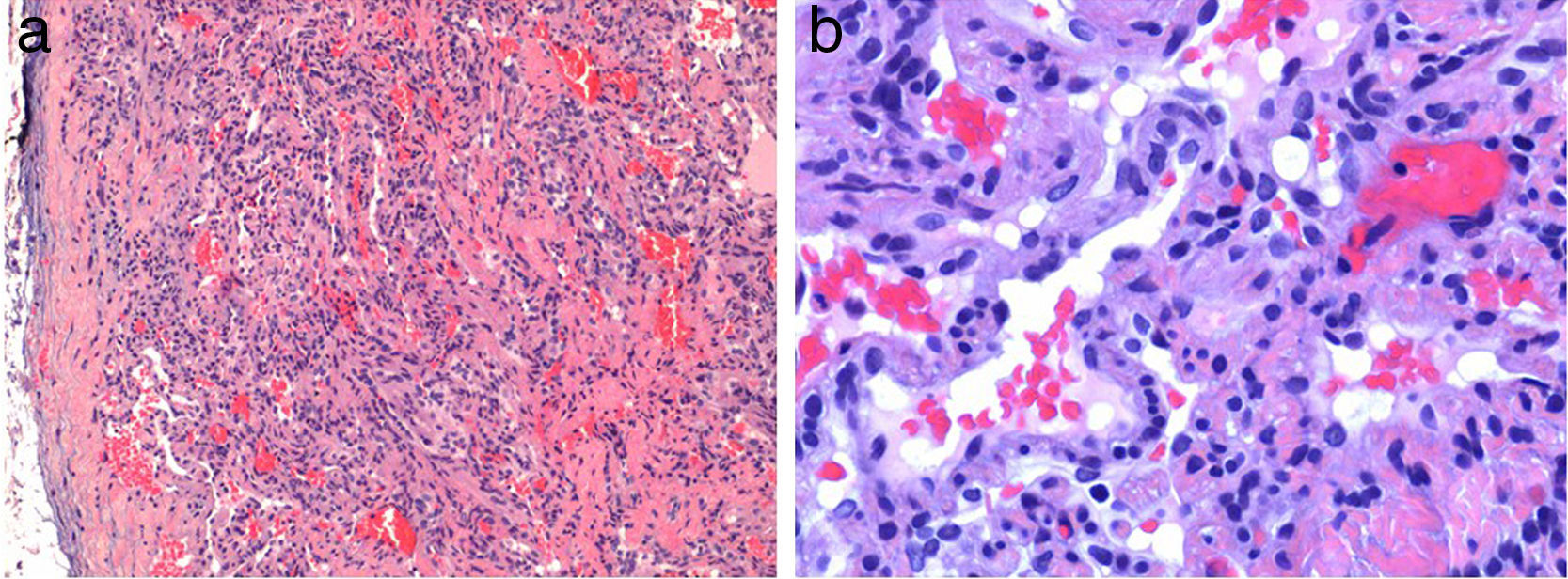
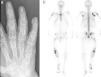
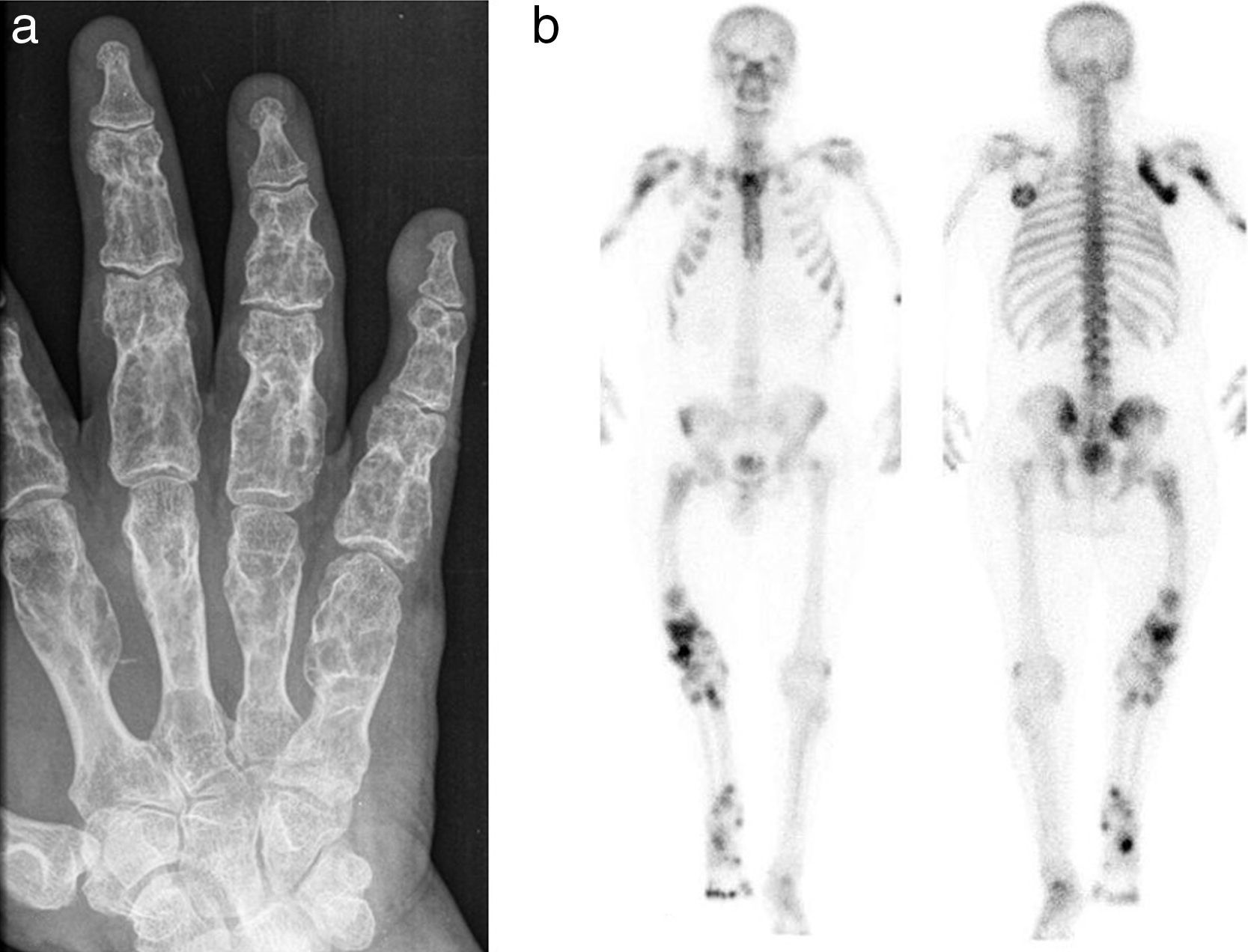
La radiografía (fig. 3a) y la resonancia magnética mostraron, tanto en las falanges como en los metacarpos, múltiples imágenes irregulares de paredes finas. La gammagrafía ósea con 99mTc-HDP puso de manifiesto múltiples focos de hipercaptación del radiotrazador, predominantemente en los huesos largos del hemicuerpo derecho (fig. 3b).
¿Cuál es el diagnóstico?
DiagnósticoSíndrome de Maffucci.
Evolución y tratamientoEl paciente sigue controles de forma periódica, y tras 2 años de seguimiento, no ha desarrollado ninguna neoplasia.
ComentarioEl síndrome de Maffucci es un trastorno infrecuente, del que únicamente hay unos 200 casos publicados en la literatura, desde que se describió en 1881. Se debe a una displasia mesodérmica congénita, no hereditaria, y se caracteriza por la tríada de encondromas y lesiones vasculares múltiples, junto a anomalías músculo-esqueléticas1–6. La etiología es desconocida, aunque artículos recientes apuntan a que podría deberse a una mutación somática, poscigótica, en las enzimas isocitrato deshidrogenasa 1 y 22.
Los pacientes suelen estar asintomáticos en el momento del nacimiento, desarrollándose las lesiones en la infancia o adolescencia, con una distribución asimétrica, predominantemente en un hemicuerpo, y en la zona distal de las extremidades1–3.
Los encondromas son tumores cartilaginosos benignos, caracterizados clínicamente por ser nódulos subcutáneos, del color de la piel normal y de consistencia ósea. Aunque son lesiones benignas, pueden presentar complicaciones, como deformidades o fracturas, siendo la complicación más importante la posibilidad de transformación maligna hacia un condrosarcoma, lo cual ocurre hasta en un 40% de los casos. Desde el punto de vista radiológico, se observan imágenes irregulares, de paredes finas insufladas, pero no destruidas; y se visualizan como áreas hipercaptantes en la gammagrafía ósea. En relación al tratamiento, en la mayoría de las ocasiones no suele ser necesario, salvo cuando aparecen complicaciones, en las que la cirugía es la opción terapéutica de elección1–6.
Con respecto a las lesiones vasculares, se presentan clínicamente como nódulos subcutáneos azulados o violáceos y de consistencia blanda. Se diferencian 3 tipos: las malformaciones venosas, las malformaciones linfáticas y el hemangioma de células fusiformes (HCF), siendo este el más característico1–6. El HCF es una proliferación vascular benigna, que a nivel histológico está constituida por 3 componentes: canales vasculares dilatados, fascículos de células fusiformes y células redondeadas con citoplasma vacuolado3,4. El riesgo de malignización de las lesiones vasculares no está establecido, aunque parece que es infrecuente. De hecho, solo hay 7 casos descritos de transformación maligna, en concreto 6 angiosarcomas y un linfangiosarcoma, aunque algunos de ellos se desarrollaron sobre zonas de radioterapia, por lo que se desconoce el papel que esta pudo tener en su aparición4. En cuanto al tratamiento, solo se requiere en casos seleccionados, siendo de elección la cirugía, a pesar de lo cual son frecuentes las recurrencias. En los últimos años se han publicado casos tratados con rapamicina, con resultados variables5.
Además del riesgo de malignización de los encondromas y las lesiones vasculares, se ha visto que los pacientes con síndrome de Maffucci pueden asociar otras neoplasias, como astrocitomas o adenomas hipofisarios6.
Por todo ello, aunque no existen protocolos establecidos de seguimiento, resulta fundamental una vigilancia estrecha de estos pacientes con el fin de detectar precozmente una posible neoplasia.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


