




Mujer de 42 años que consulta por la presencia desde hacía 5 años de lesiones nodulares agrupadas en la región dorsal izquierda, éstas habían permanecido estables y resultaban discretamente pruriginosas. Al interrogar a la paciente acerca de sus antecedentes personales y familiares, refería que había sido histerectomizada a los 32 años por miomatosis. De sus 6 hermanos (2 varones y 4 mujeres), un varón presentaba lesiones cutáneas similares pero generalizadas en el tronco y los miembros. Se había histerectomizado a una de las hermanas y a la madre por miomatosis uterina.
Exploración físicaEn la región dorsal izquierda se apreciaban múltiples nódulos de entre 2 y 5mm, eritematosos, semiesféricos, de consistencia firme, adheridos a planos profundos y que se agrupaban formando una placa de 10×4cm de diámetro (fig. 1). Tras la exploración cutánea completa no se evidenciaron otras lesiones.
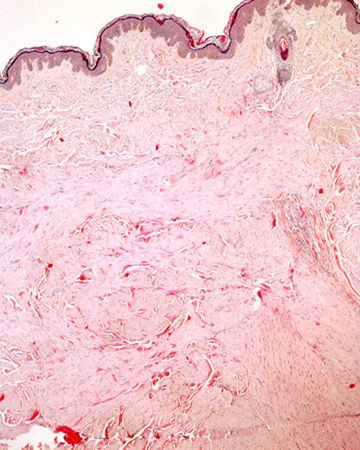
Histopatología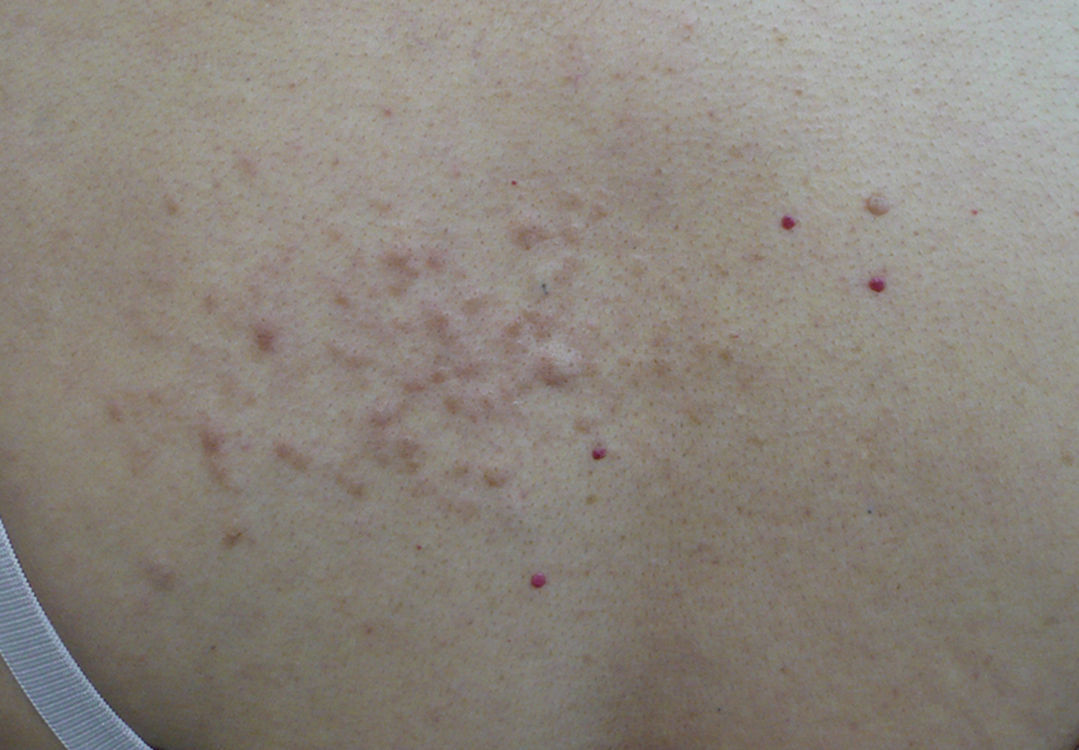
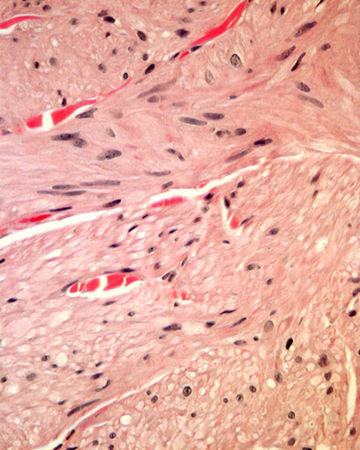
La biopsia de una de las lesiones nodulares mostró abundantes fascículos entrecruzados compuestos por células fusiformes, con núcleos centrales alargados y de bordes romos en la dermis media y profunda (figs. 2 y 3).
Los estudios de laboratorio con hemograma, bioquímica y marcadores tumorales eran normales. Se realizó una ecografía abdominal en la que no se observaron alteraciones significativas.
DiagnósticoLeiomiomatosis cutánea y uterina múltiple familiar (síndrome de Reed).
Evolución y tratamientoLas lesiones ocasionaban únicamente discreto prurito y, dada su benignidad, se optó por no tratar. La paciente acude anualmente a nuestra consulta para controlar la evolución de las lesiones cutáneas y realizar el despistaje de los posibles tumores asociados.
ComentariosEl cuadro clínico que combina leiomiomas cutáneos múltiples familiares o hereditarios con leiomiomas uterinos se denomina leiomiomatosis cutánea y uterina múltiple familiar o síndrome de Reed. Se trata de una alteración de herencia autosómica dominante con penetrancia incompleta producida por mutaciones heterocigotas en la línea germinal del gen 1q42.3-43. Éste codifica la fumarato hidrasa, enzima mitocondrial del ciclo de Krebs que cataliza la conversión de fumarato en malato y actúa como supresor de tumores1. Entre el 1–14% de los individuos con mutación del gen fumarato hidrasa asocia además carcinoma renal. Se trata de la variante conocida como leiomiomatosis hereditaria y cáncer renal2. El carcinoma renal suele corresponder a un carcinoma quístico o papilar que se desarrolla en la tercera o cuarta década de la vida y generalmente presenta un curso agresivo con metástasis tempranas3.
El piloleiomioma es un tumor dérmico benigno que deriva del músculo liso erector del pelo y puede ser solitario o múltiple. Los piloleiomiomas múltiples aparecen generalmente en el tronco con una disposición agminada, lineal o metamérica y se presentan como pápulas o nódulos semiesféricos, de entre 1 y 20mm, duros, fijos a planos profundos, de superficie lisa y color pardo rojizo, que pueden coalescer y llegar a formar placas de superficie abollonada. Con frecuencia se asocian a dolor desencadenado por frío, roce suave, presión, trauma y emociones. La evolución habitual de estos tumores es un aumento progresivo en el número y el tamaño de las lesiones, que quedan confinadas al lugar inicial de aparición. En el diagnóstico diferencial deben considerarse varias tumoraciones dérmicas como dermatofibromas múltiples agrupados, schwannomas, neurofibromas, tumores anexiales y metástasis4.
Histológicamente se observan tumores dérmicos no encapsulados con una banda subepidérmica respetada. Se componen de fascículos entrelazados formados por células fusiformes, de citoplasma eosinófilo, con núcleos centrales alargados y de bordes romos (en forma de cigarro de puro) y con vacuolas perinucleares4.
El tratamiento de las lesiones cutáneas se basa en la sintomatología o la preocupación estética. Las medidas que se requieren son, por lo general, el camuflaje cosmético y evitar los factores desencadenantes del dolor. Otra opción es la extirpación quirúrgica, aunque las recidivas son frecuentes. Se han probado tratamientos con bloqueantes-α (fenoxibenzamina), antagonistas del calcio (nifedipino), nitroglicerina, gabapentina, analgésicos y recientemente se ha publicado un caso tratado con toxina botulínica5–7. Sea cual sea la opción terapéutica elegida, lo más importante es alertar al paciente del riesgo de desarrollo de carcinoma renal y realizar un seguimiento mediante pruebas de imagen de forma periódica.
El caso que presentamos se parece tanto en la clínica como en la histología a lo clásicamente descrito en el síndrome de Reed. No pautamos tratamiento por decisión de la paciente pero sí continuamos con el seguimiento para descartar tumores asociados.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


