



HISTORIA CLÍNICA
Un varón de 33 años refería antecedentes de traumatismo hacía 2 años, y había sido diagnosticado y tratado de quiste y hematoma calcificado de rodilla. Estaba siendo estudiado por una masa en extremidad inferior izquierda. En la exploración se palpaba una masa de consistencia fibrosa y de 5 cm de diámetro, adherida a planos profundos. El resto de la exploración física no revelaba ningún dato significativo. No presentaba adenopatías ni visceromegalias. Dados los síntomas que presentaba el paciente se decidió llevar a cabo estudio y tratamiento con extirpación quirúrgica de dicha masa para informe anatomopatológico e inmunohistoquímico de la pieza (fig. 1).
EXPLORACIÓN FÍSICA
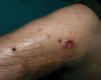
Seis meses después de la extirpación de esa masa, el paciente acudió a consulta por unos nódulos que habían aparecido en la zona próxima a la cicatriz. A la exploración se observaban 7-10 nódulos de tamaños entre 0,5 y 2 cm de diámetro, de color eritematoso pardusco, consistencia fibrosa, casi pétrea, no adheridos a planos profundos (fig. 2). El más grande de estos nódulos presentaba ulceración central. No presentaba síntomas acompañantes.
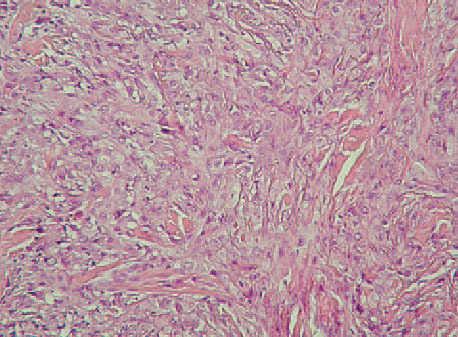
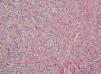
Fig. 1.Imagen anatomopatológica. (Hematoxilina-eosina, ×100.)
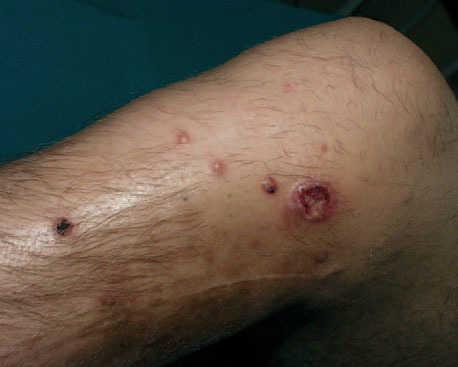
Fig. 2.Imagen clínica de la recidiva local. Nódulos de consistencia firme próximos a cicatriz de extirpación previa.
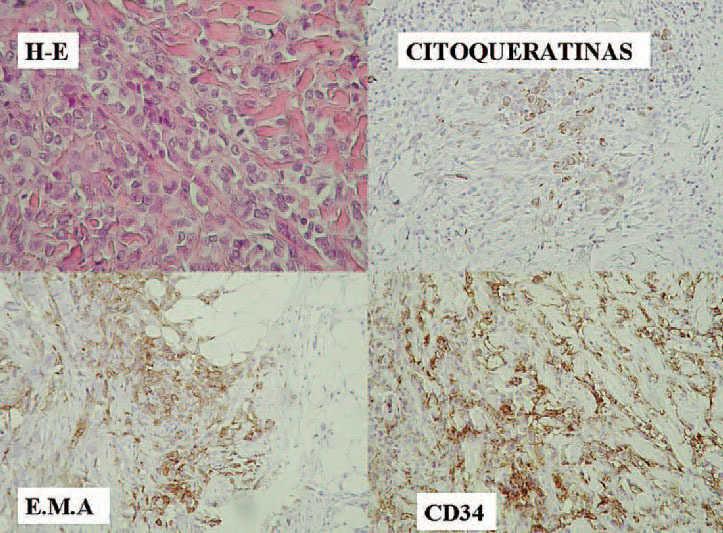
Fig. 3.Inmunohistoquímica de la lesión cutánea.
DIAGNÓSTICO
Sarcoma epitelioide (recidiva local).
HISTOPATOLOGÍA
La histopatología mostraba piel con la dermis e hipodermis infiltradas por una neoplasia no encapsulada, de bordes estrellados e infiltrantes, constituida por células fusiformes y epitelioides, de núcleos con atipias citológicas, pequeño nucléolo ocasional, y citoplasma eosinófilo amplio o claro, de límites netos. Se disponían formando nidos, cordones o nódulos, in-completamente delimitados y separados por tabiques fibroconjuntivos, con necrosis central y células epitelioides rodeándolos, que le conferían un aspecto granulomatoso, con una extensión de aproximadamente el 5 % de toda la neoplasia. Las células neoplásicas presentaban una marcada actividad mitótica, con presencia de mitosis típicas o atípicas, con un índice mitótico de 5 mitosis por 10 campos de gran aumento.
La neoplasia se encontraba bien vascularizada con vasos de calibre variable, predominantemente de tipo capilar. El inmunofenotipo de las células neoplásicas es el siguiente (fig. 3): vimentina positiva; antígeno de membrana epitelial (EMA) positivo; citoqueratinas 8, 18 y 19, positivo; citoqueratina AE, positiva, CD34, positivo. Las células son negativas para actina muscular específica (HHF-35), desmina, CD31 y proteína S-100. El índice de proliferación celular Ki 67 era de 45 %, y la expresión de proteína p53, del 35 %.
COMENTARIO
Con el diagnóstico en nuestro paciente de recidiva local de sarcoma epitelioide moderadamente diferenciado se decidió amputar la extremidad inferior hasta el tercio medio del muslo izquierdo y administrar quimioterapia complementaria con seis ciclos de adriamicina e ifosfamida. Dos meses después de finalizar el tratamiento se observaron nuevos nódulos subcutáneos en el cuero cabelludo y una adenopatía en la región supraclavicular derecha. El estudio de extensión y la punción de uno de esos nódulos resultó compatible con la neoplasia previa. Tras el diagnóstico de recidiva cutánea ganglionar, el paciente está pendiente de ser valorado nuevamente por los servicios de oncología y cirugía.
El sarcoma epitelioide es una neoplasia de tejidos blandos que fue descrita por primera vez en 1970 por Enzinger1 . La mayor incidencia se da en adolescentes y adultos jóvenes, con una relación varón:mujer de 2:1; sólo el 5 % se diagnostica en niños2 . Suele manifestarse en la parte distal de extremidades inferiores, y es el más común de los sarcomas de tejidos blandos de mano y muñeca3 . Se han descrito en casos localizados en lengua4 , vulva y pene. Afecta a la piel en el 25 % de los casos5 . La sintomatología que presenta es un nódulo único o múltiple de lento crecimiento y asintomático. Cuando se sitúa en dermis puede ulcerarse. Existe un retraso entre el primer síntoma de la enfermedad y la primera extirpación quirúrgica de 8 años de media6 , hecho éste que ensombrece el pronóstico. Las metástasis más frecuentes son ganglionares y pleuropulmonares; pero se han descrito cerebrales, óseas, pancreáticas, hepáticas, renales, cutáneas (principalmente en cuero cabelludo), tiroideas e intestinales. Suelen presentarse como más de un nódulo como media a los 5 años del diagnóstico. El diagnóstico diferencial clínico incluye verruga vírica, granuloma específico e inespecífico, dermatofibroma, tenosinovitis, nódulo reumatoide, ganglión; cuando existe ulceración, el diagnóstico diferencial debe establecerse con el carcinoma espinocelular.
Histológicamente está definido por una proliferación de células fusiformes, poliédricas, epitelioides, de citoplasma muy eosinófilo y núcleo grande. Estas células se disponen formando nódulos bien definidos no encapsulados en la dermis y la hipodermis, centrados por zonas de necrosis. Es por esta disposición, que recuerda a los granulomas, que desde el punto de vista anatomopatológico puede confundirse con múltiples enfermedades de naturaleza benigna inflamatoria o infecciosa como los granulomas anular, sarcoideo o tuberculoso, la necrobiosis lipoídica, el nódulo reumatoideo, la fascitis nodular o la tenosinovitis. También puede confundirse con procesos tumorales como carcinomas escamosos y anexiales, melanoma y tumores malignos vasculares epitelioides. Algunos sar-comas epitelioides presentan células rabdoides grandes de gran atipia citológica, con citoplasma eosinófilo y núcleo excéntrico, por lo que debe el diagnóstico diferencial plantearse con el tumor maligno rabdoide extrarrenal. Presenta dificultad diagnóstica al asemejarse a otros sarcomas de aspecto pseudoepitelial como schwannoma maligno epitelial, hemangioendotelioma epitelioide y, sobre todo, con el sarcoma sino-vial, más aún si profundiza en dermis e hipodermis y se adhiere a fascia o tendón.
Se han descrito variantes histopatológicas del sarcoma epitelioide como la rabdoide, la fibromatosa y la angiomatoide7 . En esta última se describen espacios quísticos de contenido hemorrágico, semejantes a los que aparecen en el angiosarcoma. La forma rabdoide está constituida por grandes células rabdoides como las que se describen en los tumores rabdoides de tejidos blandos que se han referenciado anteriormente. Estas formas son muy características de las localizaciones proximales. Por último, las formas fibromatosas aparecen sobre todo en momentos evolutivos de las fases tempranas del tumor. Son las que pueden confundirse con procesos fibrosos de naturaleza benigna como el histiocitoma fibroso y la fibromatosis. La inmunohistoquímica es de gran ayuda en el diagnóstico diferencial, y muestra coexpresión de queratina y vimentina; la proteína S-100, CD31 y HMB-45 son generalmente negativas.
El tratamiento de elección es la cirugía radical, dado que es poco radio y quimiosensible. Si el tumor es mayor de 5 cm o la escisión no puede ser lo suficientemente amplia se realizará radioterapia complementaria. En pacientes con metástasis o enfermedad residual importante se aconseja tratamiento con quimioterapia, aunque no existe evidencia de que mejore la supervivencia 8 . Son factores de mal pronóstico el sexo masculino, ser mayor de 40 años, la localización en tronco o parte proximal de extremidades, la invasión vascular, el tamaño del tumor mayor de 5 cm, más de 30 % de necrosis, y un alto índice mitótico. A los 5 años, la media de supervivencia es del 70 % 9 .
Correspondencia:
María Huerta. Secretaría de Dermatología. Máiquez, 7. 28007 Madrid. España. mhbrogeras@hotmail.com
Recibido el 15 de marzo de 2004. Aceptado el 4 de junio de 2004.


