


Además de las manifestaciones dermatológicas diagnósticas de la NF1 (MCCL, efélides y neurofibromas), existen otras alteraciones cutáneas como los nevus anémicos y los xantogranulomas juveniles, cuya presencia parece ser de gran valor predictivo en los pacientes sin diagnóstico de certeza. La NF1 mosaica localizada, clásicamente denominada neurofibromatosis segmentaria, se produce a causa de una mutación poszigótica que determina que la presencia de las manifestaciones típicas de la enfermedad se limite a un segmento corporal. Aunque no existen protocolos de seguimiento de estos pacientes, el riesgo de complicaciones es mucho menor que en las formas generalizadas de la enfermedad. Los tumores malignos son, probablemente, la complicación más temida de la NF1. En orden decreciente, las neoplasias más frecuentes son los gliomas del nervio óptico, el tumor maligno de vaina nerviosa periférica (TMVNP), los tumores estromales gastrointestinales y los feocromocitomas. La mayoría de los TMVNP son el resultado de la transformación maligna de los neurofibromas plexiformes y su diagnóstico precoz es determinante para el pronóstico. En conclusión, la NF1 es una enfermedad multisistémica que requiere seguimiento multidisciplinar, pero en cuyo diagnóstico y seguimiento los dermatólogos tenemos un papel destacado.
Neurofibromatosis type 1 (NF1) is the most common neurocutaneous syndrome and probably the one best known to dermatologists. Although the genetic locus of NF1 was identified on chromosome 17 in 1987, diagnosis of the disease is still based primarily on clinical observations. The 7 diagnostic criteria of the National Institutes of Health, which were established in 1988, include 3 skin manifestations (café-au-lait spots, freckling on flexural areas, and cutaneous neurofibromas). The age at which these diagnostic lesions appear is variable: onset can be late in some patients while others never develop certain symptoms. Definitive diagnosis may therefore be delayed by years. Although the appearance of the characteristic café-au-lait spots and freckling in the early years of childhood are very suggestive of the disease, these signs are not pathognomonic and, in isolation, do not constitute sufficient evidence to establish a definitive diagnosis. Thus, other diagnoses should be considered in patients whose only symptoms are café-au-lait spots and freckling. By contrast, the presence of multiple cutaneous neurofibromas or at least 1 plexiform neurofibroma is a very specific indication of NF1. Identification of the different types of neurofibroma allows us to confirm the diagnosis and initiate appropriate management.
Además de las manifestaciones dermatológicas diagnósticas de la NF1 (MCCL, efélides y neurofibromas), existen otras alteraciones cutáneas muy frecuentes (tabla 1), algunas de las cuales pueden tener un importante valor predictivo en los niños con MCCL típicas sin diagnóstico definitivo de NF1. Por otro lado, la implicación de la neurofibromina en la vía RAS-MAPK va a interferir con la proliferación y diferenciación celular y, por tanto, va a aumentar la predisposición al desarrollo de tumores malignos en estos pacientes.
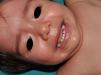
Otras manifestaciones cutáneas frecuentes en la neurofibromatosis tipo 1Nevus anémicosLa asociación entre NF1 y los nevus anémicos (NA) fue sugerida por Naegeli en el año 19151, pero apenas recibe atención en la literatura hasta el año 20132–5. Los NA son máculas pálidas de contorno polilobulado cuyo tamaño oscila entre pocos milímetros a varios centímetros; son muy sutiles desde el punto de vista clínico, poniéndose de manifiesto al frotar ligeramente la zona (fig. 1). Las lesiones pueden ser únicas o múltiples y localizarse en cualquier zona del cuerpo, aunque son más frecuentes en la región preesternal3,5. Si bien las series publicadas reflejan una elevada prevalencia en los niños, no se conoce con precisión el momento de aparición de las lesiones, ya que con mucha frecuencia los padres no se han percatado de la existencia de los NA hasta que nosotros los detectamos en la consulta. En nuestra serie de pacientes los NA estaban presentes en al menos el 50% de los niños con NF1 confirmada5. Probablemente, las diferencias de prevalencia entre los distintos estudios, que oscilan entre el 8,8% y el 51%3, estriban en su naturaleza retrospectiva o prospectiva respectivamente, ya que son difíciles de diagnosticar salvo que se busquen de manera sistemática. Para detectarlos, nosotros preguntamos rutinariamente a los padres si han visto lesiones blanquecinas transitorias durante el baño, los procesos febriles, el llanto o el ejercicio, y frotamos a ciegas todo el área preesternal en los bebés y los niños pequeños para resaltar las lesiones. Los NA parecen deberse a la ausencia de respuesta focal a la acción de las catecolaminas, e histológicamente son indistinguibles de la piel normal.
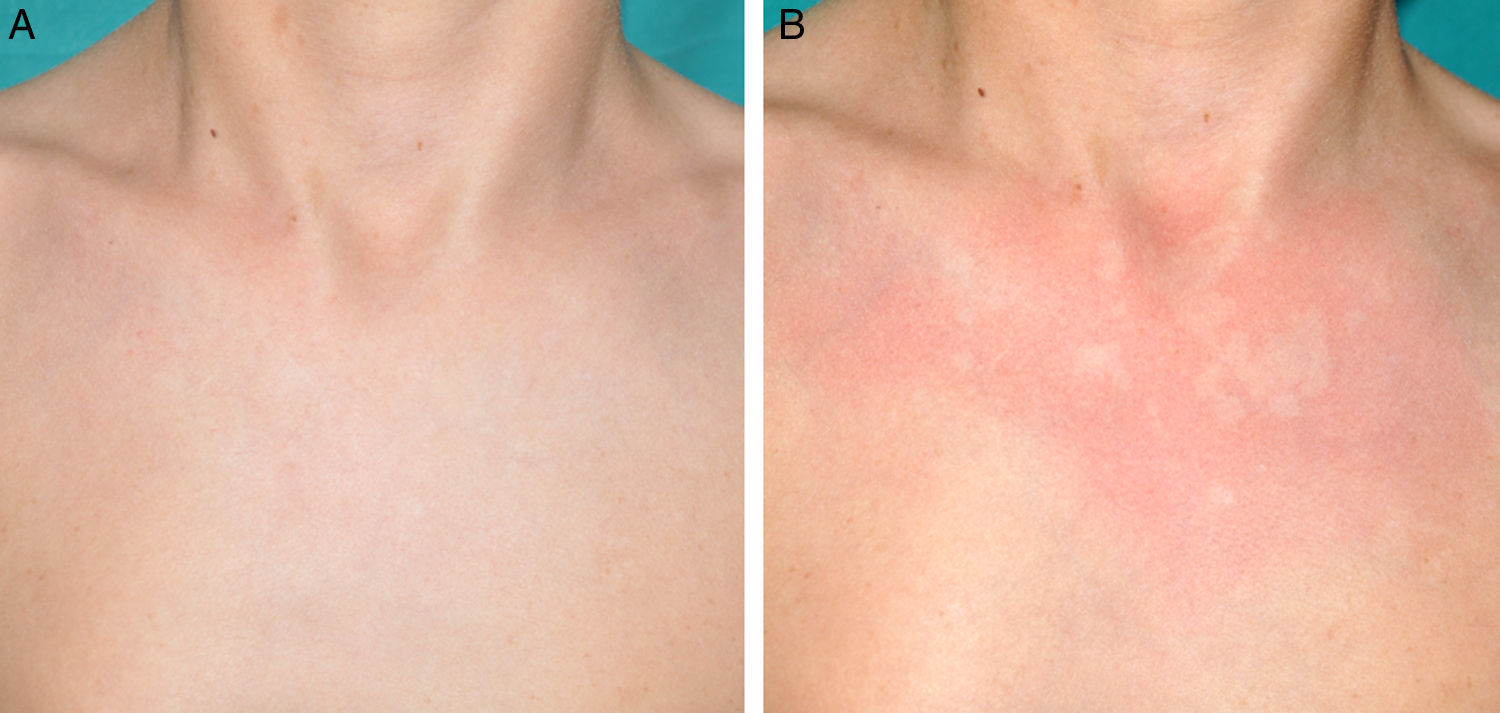
Aunque los NA pueden aparecer en individuos sanos, particularmente en proximidad con malformaciones capilares, no se han descrito en asociación con ninguna otra genodermatosis con MCCL ni con la neurofibromatosis segmentaria. De acuerdo con otros autores2–4, nosotros pensamos que los NA son un hallazgo distintivo en la NF1 y que su presencia tiene un importante valor predictivo de NF15.
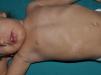
Xantogranulomas juvenilesLos xantogranulomas juveniles (XGJ) son la forma más frecuente de histiocitosis de células no Langerhans, y un hallazgo relativamente común en la NF16 (fig. 2). Las lesiones desaparecen espontáneamente al cabo de pocos años, siendo raro encontrarlas en la infancia tardía y la edad adulta7. Su prevalencia estimada en pacientes con NF1 oscila entre el 0,7% en adultos8 y el 37,5% en niños7, siendo la cifra particularmente elevada en niños menores de 9 años4,7. En nuestra serie la prevalencia de XGJ en pacientes pediátricos con diagnóstico de certeza de NF1 fue del 6,2%5, cifra más parecida al 8,5% observado por otros autores3. En todo caso, es innegable que se trata de un hallazgo muy significativo en los pacientes con NF1, y que puede ser de particular trascendencia en los casos sin diagnóstico de certeza. Así, un estudio observó que en 8 de 10 pacientes con NF1 que solo tenían MCCL en la primera visita y que en la exploración presentaban XGJ o nevus anémicos, desarrollaban la enfermedad con posterioridad4, y otro encontró que en todos los niños que tenían XGJ y MCCL se confirmaba luego la NF17. Por tanto, considerando que el XGJ es un tumor muy frecuente en la NF1, es probable que la coexistencia de MCCL y XGJ en niños pequeños no deba considerarse un hecho casual, sino un hallazgo altamente indicativo de la enfermedad. Su presencia también ha sido relacionada con un mayor riesgo de leucemia infantil en los pacientes con NF1, en particular de leucemia mielomonocítica juvenil (LMMJ)9. Sin embargo, esta asociación no ha sido refrendada por otros estudios, y ha sido cuestionada por algunos autores10–12, en particular dentro de la literatura dermatológica. Aunque no faltan casos esporádicos en la literatura con dicha asociación13–16, no se ha observado una mayor incidencia de LMMJ en largas series de pacientes con NF1 que presentaron XGJ, incluyendo la nuestra4,5,7. Por tanto, la sospecha de LMMJ en un paciente con NF1 que desarrolla XGJ debería estar dirigida por la sintomatología clínica y la exploración física, y no por el mero hallazgo11.
Tumores glómicos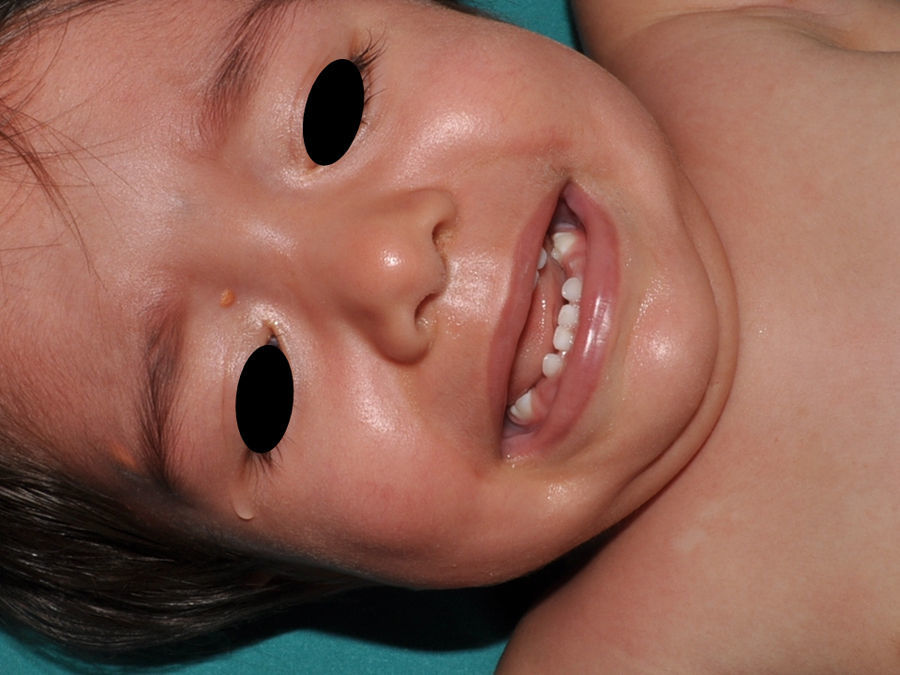
La asociación de la NF1 con los tumores glómicos se sospecha desde 193817, pero la relación quedó definitivamente establecida en 2009, cuando se demostró la inactivación bialélica del gen NF1 en las células del glomus18. Los tumores glómicos son lesiones vasculares benignas que se originan en el cuerpo del glomus, un organismo neuro-mio-arterial especializado en la regulación del flujo vascular. Suelen localizarse en zonas acrales, particularmente en la región subungueal, y se caracterizan por producir un dolor paroxístico a la presión y con los cambios de temperatura. En los pacientes con NF1 tienden a ser múltiples y recidivantes19. Son raros en los niños, estimándose una prevalencia del 5% en los pacientes adultos con NF120. Aunque pueden aparecer en individuos sanos, en torno al 30% de los pacientes con tumores glómicos sufren NF119,20. Desde el punto de vista histológico se caracterizan por la existencia de vasos de pared fina rodeados por filas de células uniformes de núcleo redondo y oscuro que, a diferencia de lo que ocurre en los casos esporádicos, en los individuos con NF1 no expresan neurofibromina19.
Otros hallazgos cutáneos frecuentes en la neurofibromatosis tipo 1El prurito, la hiperpigmentación generalizada, la presencia de máculas hipocrómicas, y la suavidad de la piel en los pacientes son hallazgos fácilmente constatables en los pacientes con NF1.
El prurito afecta al 20% de los pacientes con NF1 y altera considerablemente su calidad de vida21. El picor se ha atribuido al aumento de los mastocitos en la piel, pero no parece haber diferencias en los niveles plasmáticos de histamina en los pacientes con picor respecto a la población general22. Por otra parte, muchos pacientes también sufren prurito localizado en los neurofibromas, en los que sí se ha demostrado un número elevado de mastocitos y la inhibición del picor con la administración de ketotifeno23. No hay que olvidar que la presencia de picor localizado puede alertar sobre el desarrollo de tumores medulares o del sistema nervioso central24,25, por lo que es un síntoma relevante en los pacientes con NF1.
Los pacientes con NF1 presentan una hiperpigmentación generalizada difusa que se hace evidente al comparar el tono de su piel con el de familiares no afectados, y explica la hiperpigmentación de base que exhiben con frecuencia las formas segmentarias NF1. Recientemente se ha demostrado in vitro que los melanocitos con mutación en el gen NF1 reproducen esta hiperpigmentación, la cual se atribuye a la sobreexpresión del factor de transcripción MITF, la tirosinasa y la tautomerasa dopacroma, agentes todos ellos involucrados en la melanogénesis26.
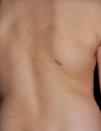
Al igual que en la esclerosis tuberosa se observan MCCL, en los niños con NF1 se pueden encontrar máculas hipopigmentadas con relativa frecuencia27 (fig. 3), pero hasta el momento no hay estudios que las hayan caracterizado en prevalencia, número y morfología. Finalmente, la suavidad de la piel de los pacientes con NF1 es una percepción subjetiva que también es resaltada por algunos expertos en la enfermedad27.
Neurofibromatosis segmentaria o mosaica localizada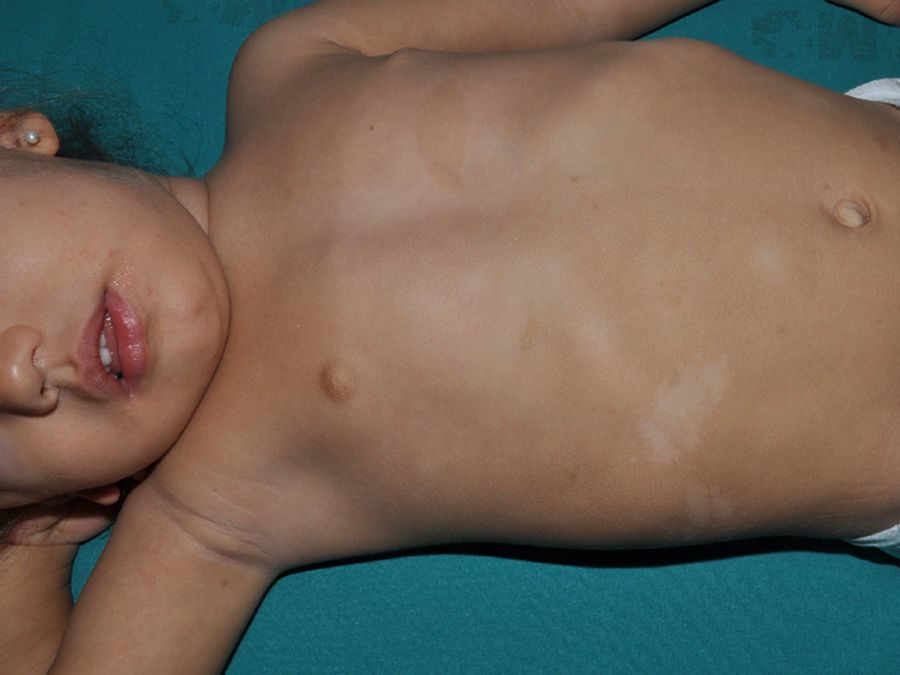
La neurofibromatosis segmentaria tiende a denominarse en la actualidad neurofibromatosis mosaica localizada (NFML). Se caracteriza porque las manifestaciones típicas de la NF1 están limitadas a uno o varios segmentos del cuerpo. Se distinguen 4 tipos distintos en función de los hallazgos clínicos: 1) NFML con cambios cutáneos pigmentarios (MCCL y efélides) exclusivamente; 2) NFML con neurofibromas solamente; 3) NFML con cambios cutáneos pigmentarios y neurofibromas; y 4) NFML con neurofibromas plexiformes exclusivamente28. No es necesario un número mínimo de MCCL, que las pecas se localicen en las flexuras para poder diagnosticar una NFML, ni tampoco que haya un número mínimo de 2 neurofibromas cutáneos en la zona de cambios pigmentarios típicos. Se estima una prevalencia de entre 1:30.000 y 1:40.000 personas sanas8, pero probablemente sea más frecuente, ya que a menudo pasa desapercibida por médicos y padres, que la consideran una «marca de nacimiento». El desarrollo de las lesiones sigue el mismo curso cronológico que las de la forma sistémica de la enfermedad. Por tanto, la mayoría de los pacientes presentarán lesiones exclusivamente pigmentarias durante la niñez y desarrollarán neurofibromas a partir de la pubertad28,29. En las formas con cambios exclusivamente pigmentarios es frecuente observar una tenue hiperpigmentación de base que delimita el segmento afectado el cual, aunque netamente definido, puede traspasar ligeramente la línea media (fig. 4). Algunos pacientes presentan MCCL fuera del segmento aparentemente involucrado, particularmente cuando el segmento afecta la raíz escapular o pélvica, casos en los pueden existir MCCL a lo largo de la extremidad involucrada. Otros hallazgos cutáneos, como los NA y los XGJ, no han sido descritos, y las complicaciones sistémicas que se asocian con frecuencia a la NF1 parecen ser menos frecuentes en la NFML30. La NFM suele ser asintomática, pero puede producir picor31 o dolor por compresión radicular o secundario a la malignización32,33.
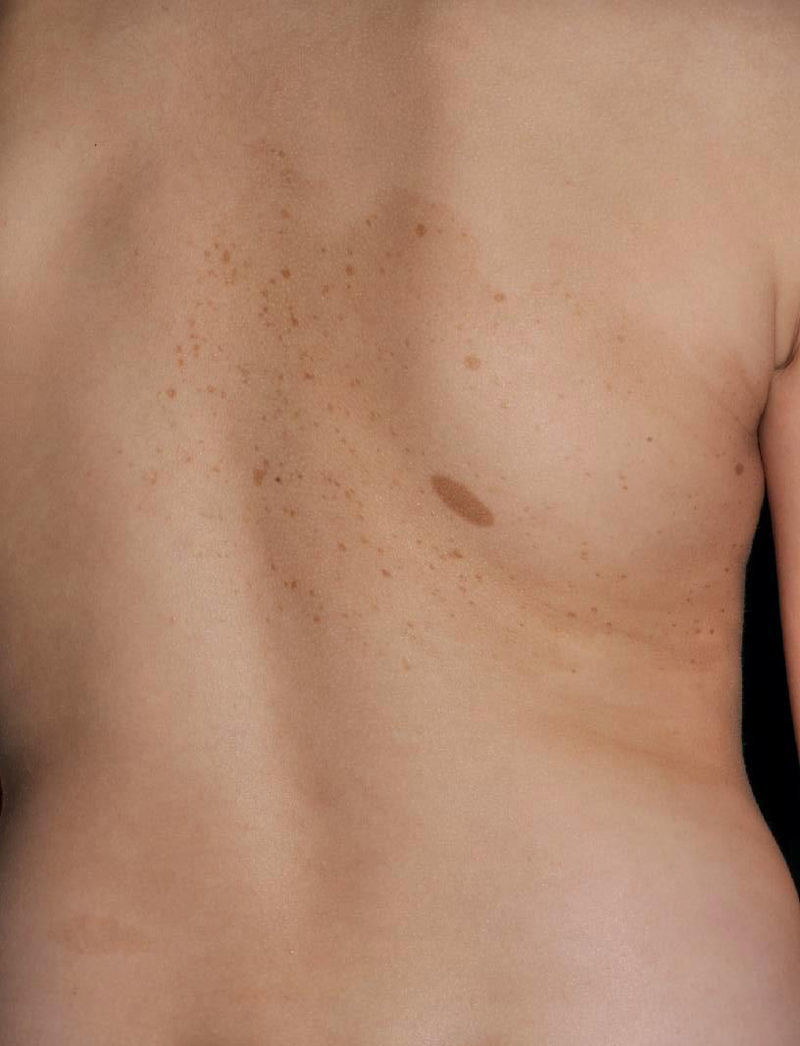
No es infrecuente que la NFML sea confundida con otras lesiones como el nevus spilus, mosaicismos pigmentarios o, muy particularmente, pigmentaciones lentiginosas segmentarias34. Estas pigmentaciones son muy difíciles de distinguir clínicamente de una NFML, y la diferenciación solo podría hacerse mediante análisis molecular tisular. Los pacientes con formas mosaicas de NF no deben ser diagnosticados de NF1 generalizada, aun cuando cumplan criterios diagnósticos por el número y tamaño de la MCCL que presentan en la zona afectada. No obstante, los pacientes deben saber que existe un pequeño riesgo de transmitir la enfermedad generalizada a los descendientes28,30. Es importante destacar que el mosaicismo gonadal es independiente de localización del área cutánea afectada, la cual no necesariamente se localiza sobre o cerca de la zona gonadal. Además, no existen datos concluyentes acerca del riesgo de padecer un mosaicismo germinal, ya que aunque en una reciente revisión sistemática se observó que 4 de 157 casos publicados (2,5%) transmitieron una forma generalizada de la enfermedad30, los propios autores asumen un sesgo de publicación. Por otro lado, no se dispone de datos sobre el número total de descendientes de los pacientes con NFML comunicados y el porcentaje de hijos afectados, una información esencial para hacer un cálculo fidedigno del riesgo de transmisión de la enfermedad.
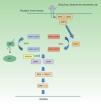
Neurofibromatosis tipo 1 y cáncerLos tumores malignos son, probablemente, la complicación más temida de la NF1. La ruta de señalización mediada por las MAP-quinasas ERK1 y 2 desempeña un papel esencial en el control de la proliferación, diferenciación y supervivencia celular en condiciones fisiológicas, por lo que los fallos acaecidos en la regulación de dicha ruta contribuyen significativamente a la transformación celular, y están incuestionablemente involucrados en la progresión tumoral (fig. 5). En orden decreciente, las neoplasias más frecuentes en la NF1 son los gliomas del nervio óptico (GVO) (15-20%), el tumor maligno de la vaina nerviosa periférica (TMVNP) (8-13%), los tumores estromales gastrointestinales (4-25%) y los feocromocitomas (0,1-5%)35. Además, sufren una probabilidad de 7 a 5 veces mayor que la población general de sufrir leucemia, tumores cerebrales y cáncer de mama. Por último, aunque no está clara la relación de la NF1 y el melanoma, recientes hallazgos involucran a la neurofibromina en la génesis de dicho tumor. Por motivos de espacio solo comentaremos los tumores de estirpe neural y la relación entre la neurofibromina y el melanoma.
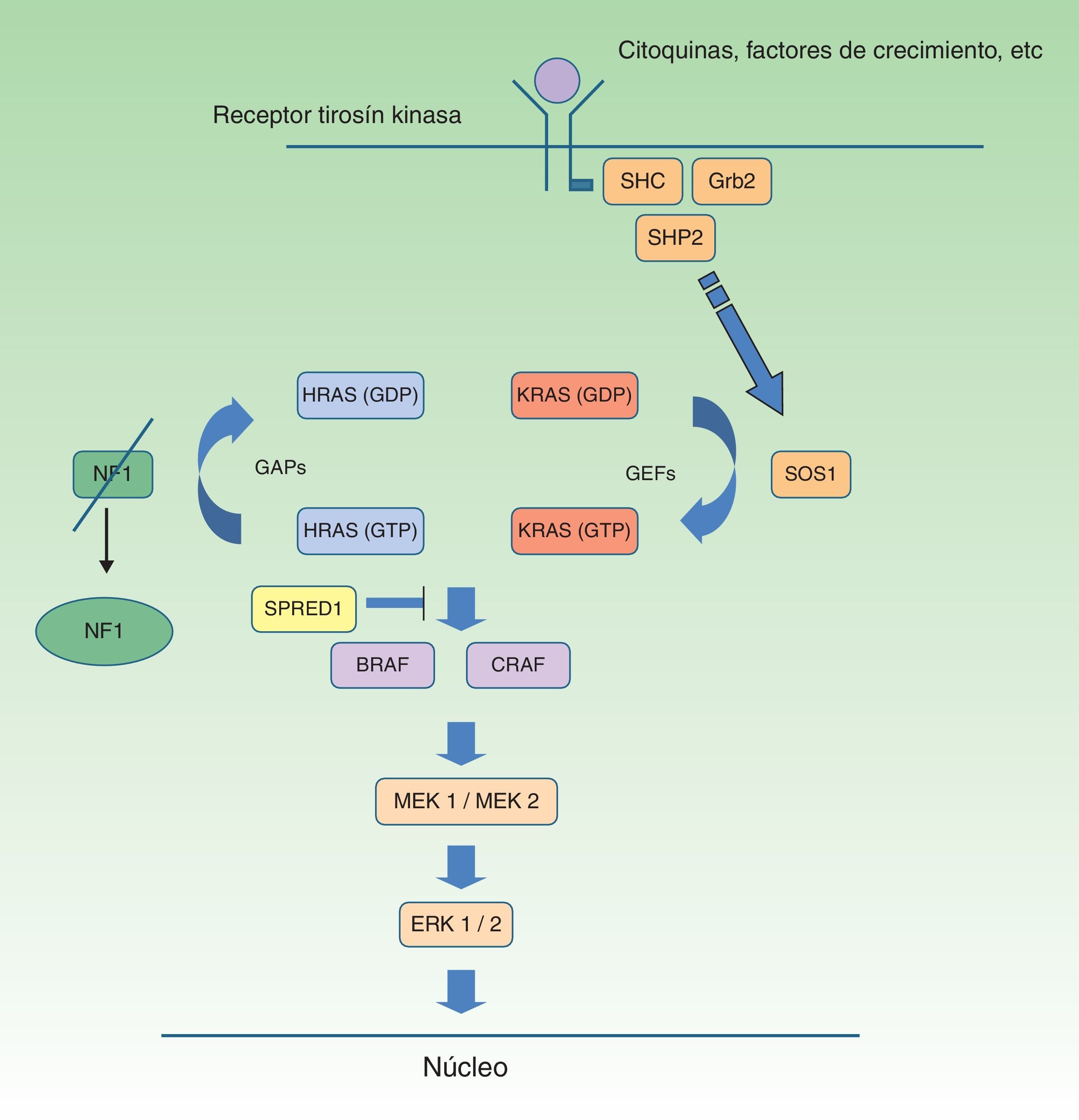
Ruta metabólica RAS MAPK. Tras la estimulación de los receptores celulares se activan proteínas intracelulares (SHC, GRB2 y SHP2), que reclutan el SOS1 intracitoplásmico, activando las proteínas RAS. La activación de las proteínas RAS se acompaña de la activación de RAF (BRAF, RAF1), MEK1A1/MEK1A2 y, por último, ERK1/ERK2, que son los últimos efectores de la vía RAS/MAPK y responsables del mantenimiento del ciclo de vida celular.
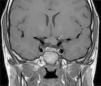
Los GVO suponen el 85% de los gliomas de bajo grado (astrocitomas pilocíticos) de los pacientes con NF1, y afectan al 15-30% de los niños entre 3 y 5 años36,37. Constituyen un criterio diagnóstico de la enfermedad. En la mayoría de los casos son asintomáticos, pero pueden causar pérdida de visión hasta en un 12% de los pacientes37 o pubertad precoz35. La elevada incidencia de los gliomas de nervio óptico hace aconsejable un estrecho seguimiento oftalmológico en los niños con NF1, ya sea clínico o clínico y radiológico, ya que la detección clínica puede ser difícil y algunos neuropediatras prefieren realizar pruebas de imagen complementarias (fig. 6)37. El 15% restante de los gliomas de bajo grado también son más frecuentes en la primera década de la vida, y asientan en cualquier parte del cerebro, manifestándose clínicamente con cefalea, marcha inestable o letargia35. Por el contrario, los gliomas de alto grado (glioblastomas multiformes) son propios de los adultos jóvenes. El riesgo de padecerlos quintuplica el de la población general y tienen mal pronóstico35,38. El tratamiento del GVO en los pacientes con NF1 suele ser conservador, salvo que se observe disminución en la agudeza visual y progresión del tamaño en las pruebas de neuroimagen. En caso de ser necesario, la primera línea de tratamiento es la quimioterapia con vincristina y carboplatino. La radioterapia se desaconseja debido al alto riesgo de tumores secundarios, y el tratamiento quirúrgico queda reservado para tumores orbitales de gran tamaño sin visión útil, con exposición de la córnea o si hay proptosis39.
Tumor maligno de la vaina nerviosa periférica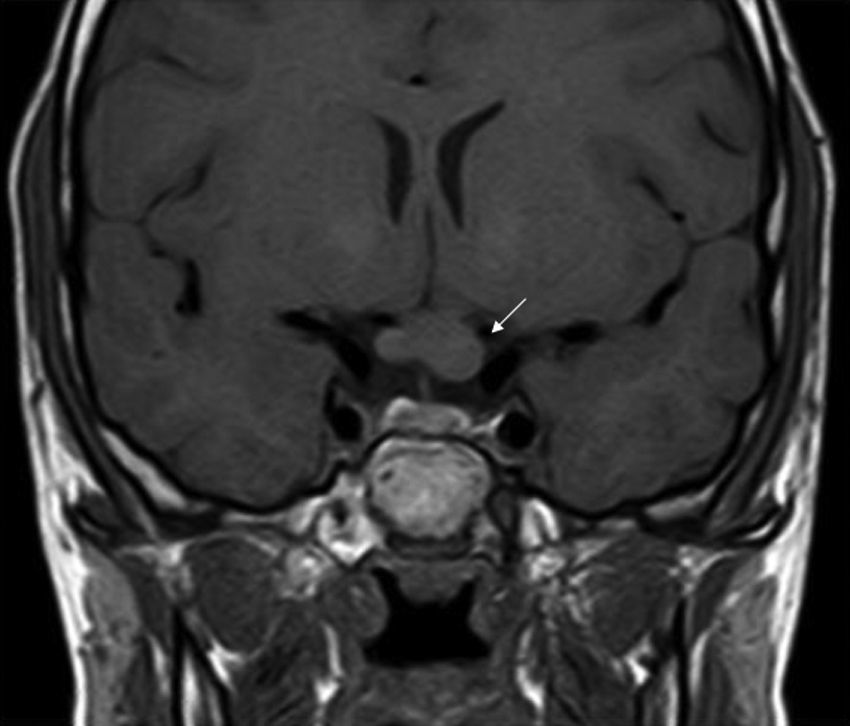
El TMVNP (o neurofibrosarcoma) es un tipo de sarcoma que se cree que deriva de la célula de Schwann. Supone el 3-10% de todos los sarcomas y casi siempre está relacionado con la NF1, en cuyos pacientes tiene una prevalencia aproximada del 0,1%, y un riesgo acumulado a lo largo de la vida del 8-13%35. En la NF1 se presentan en<5% de los pacientes adultos40,41 y en menos del 2% de los niños36, siendo el riesgo mayor en las áreas con un neurofibroma plexiforme preexistente, cuando hay deleción completa del gen o si hay antecedentes de radioterapia previa. Los factores predictivos de la existencia de neurofibromas plexiformes internos (o profundos) han sido comentados en otro apartado, pero aunque la mayoría de los TMVNP son el resultado de la transformación maligna de los neurofibromas plexiformes, también pueden aparecer sobre neurofibromas subcutáneos e incluso sin lesión previa sin lesión previa36,40,42. No se conocen bien los mecanismos moleculares de la transformación maligna de los neurofibromas, pero la mutación bialélica del gen NF1 en los tumores, junto con otros mecanismos epigenéticos, puede llevar a la mutación de otros genes importantes como son p53 e InK4A41. Los síntomas de alarma incluyen aumento brusco de tamaño, cambio de textura (aumento de la consistencia), dolor intenso o incoercible y sintomatología neurológica distinta de la habitual40. En estos casos es obligada una evaluación radiológica, una biopsia y, en su caso, la resección quirúrgica inmediata, ya que el retraso en el diagnóstico (en directa relación con el tamaño del tumor) se correlaciona directamente con el pronóstico43. Hay que tener en cuenta que la RMN ofrece datos sobre la extensión y localización, pero no sobre el comportamiento biológico del tumor, por lo que para valorarlo es necesario realizar una PET con 18 fluorodesoxiglucosa, que también dará información sobre posibles metástasis. No es conveniente realizar una PAAF, ya que el estudio histológico debe contener una muestra suficiente de tejido. La cirugía radical es la única alternativa terapéutica curativa, pero a menudo es imposible o demasiado mutilante. El tratamiento quimioterápico coadyuvante es controvertido y no es estándar, salvo en el caso de la antralina en pacientes con tumor metastásico44,45. En la actualidad el TMVNP tiene un pobre pronóstico de supervivencia35.
MelanomasNo está claro si existe una verdadera asociación entre NF1 y melanoma o los casos publicados, la mayoría aislados, son anecdóticos. En la serie más larga que existe, que incluyó 11 pacientes con NF1 y melanomas cutáneos, se observó que este tumor era más frecuente en mujeres (en proporción 1:10), fundamentalmente jóvenes (mediana de edad=33) y que el Breslow medio era de 3,2mm, considerablemente mayor que en otras series de pacientes con melanoma sin NF146, quizá porque el resto de las lesiones pigmentadas dificulta la detección. A pesar de esta incierta asociación, la relación entre el melanoma y la vía RAS es irrefutable, ya que no solo el 50% de los melanomas exhiben mutaciones en BRAF (particularmente V600E) y un 15-20% de los mismos tienen mutaciones en NRAS (sobre todo en el codón 61), sino que muchos melanomas presentan mutaciones en NF1 asociadas o no a las anteriores47, lo que le convierte en el tercer gen mutado con mayor frecuencia en el melanoma. Este hallazgo parece particularmente frecuente en los melanomas desmoplásicos, donde se detectan mutaciones del gen NF1 en el 93% de los casos, frente al 20% de los melanomas no desmoplásicos48. Sin embargo, que la presencia de estas mutaciones tenga valor predictivo en la respuesta al tratamiento con inhibidores MEK es controvertido49,50. Además de en el melanoma, se han detectado mutaciones del gen de la neurofibromina en otros cánceres esporádicos como tumores cerebrales, de mama, ováricos y leucemias, lo cual está abriendo prometedores horizontes terapéuticos51.
Finalmente, es importante destacar que la tasa de mortalidad por cáncer en los individuos con NF1 es algo mayor en individuos menores de 50 años, fundamentalmente a expensas del cáncer de mama y la LMMJ, pero se iguala con la de la población general a partir de esa edad52. Del mismo modo, aunque la esperanza media de vida es algo menor de lo esperado en los individuos sanos, la mayoría de los pacientes con NF1 sobrepasan los 70 años.
Diagnóstico de la neurofibromatosis tipo 1En la actualidad el diagnóstico de la NF1 sigue siendo fundamentalmente clínico53. Sin embargo, el desarrollo cronológico de los criterios diagnósticos dificulta el diagnóstico en los niños pequeños, y de hecho solo un 46% de los pacientes sin antecedentes familiares de la enfermedad fueron diagnosticados antes de los 2 años de vida54. En los últimos años diversos autores han propuesto incluir otros hallazgos cutáneos como los nevus anémicos y los xantogranulomas entre los criterios diagnósticos para facilitar el diagnóstico en este grupo de edad2–5,7,27, y algunos incluso reclaman la revisión y actualización de los criterios clínicos clásicos ya27,55.
El diagnóstico genético de la NF1 no es de acceso generalizado. El gran tamaño del gen NF1, la existencia de pseudogenes homólogos en regiones pericéntricas de otros cromosomas, la escasa correlación genofenotípica y la ausencia de zonas mutacionales calientes o hotspot hacen que el estudio mutacional sea laborioso y caro. La combinación de técnicas moleculares aumenta la sensibilidad de la detección de las anomalías genéticas. En concreto, la combinación del análisis de ARN (denaturing high performance liquid chromatography) y multiplex ligation-dependent probe amplification, validada por autores españoles, tiene una sensibilidad del 95% en la detección de mutaciones del gen NF1 y proporciona resultados muy fiables56. Hasta el momento se han descrito más de 1.000 mutaciones a lo largo de todo el gen NF1; la mayoría de las mutaciones en NF1 se producen por cambios puntuales en la secuencia codificante del gen, y solo una minoría de casos presenta una deleción genética. Aunque la penetrancia de la enfermedad es prácticamente del 100% en los adultos, en muy pocos casos hay una buena correlación genofenotípica, ni siquiera dentro de la misma familia o en gemelos idénticos57-60. Las excepciones son los casos de microdeleción completa del gen NF1 de 1.4Mb (en la que los pacientes sufren una forma grave de la enfermedad, presentando neurofibromas numerosos y precoces, anomalías cognitivas, rasgos dismórficos y tendencia a la malignización)58,61, las deleciones de 3 pares de bases en el exón 17 afectando a un único aminoácido, p.Met992del (casos que presentan MCCL y efélides y ausencia de neurofibromas)60 y las mutaciones en el codón 1809 del exón 29, que cursa con estenosis pulmonar y baja talla, características fenotípicas del síndrome de Noonan59.
La dificultad del estudio genético hace que algunos autores consideren innecesario el estudio genético de rutina en los niños pequeños que todavía no cumplen criterios de la enfermedad, ya que: 1) el 95% de los niños cumplirá criterios clínicos a los 8 años de vida; 2) un resultado negativo en la prueba genética, en ausencia de una mutación familiar conocida, no excluye el diagnóstico; y 3) la ausencia de correlación genofenotípica impide avanzar un pronóstico y no modifica el seguimiento62,63. En contra de esta opinión está la realidad cotidiana de los médicos que atendemos a los niños con NF1: unos padres angustiados (y perfectamente informados sobre los riesgos y peores perspectivas de la enfermedad) que quieren saber con certeza si su hijo sufre o no la enfermedad. Presumiblemente, los avances tecnológicos en el diagnóstico molecular y el abaratamiento de los costes facilitarán muy pronto la generalización del estudio genético.
En conclusión, la presencia de hallazgos cutáneos como los NA y los XGJ parecen ser de gran valor predictivo en los pacientes con MCCL sin diagnóstico de certeza de NF1. Los enfermos con NF1 tienen predisposición al cáncer, siendo el diagnóstico precoz de los tumores malignos determinante para el pronóstico. La NF1 es una enfermedad multisistémica que requiere seguimiento multidisciplinar, pero en cuyo diagnóstico y seguimiento los dermatólogos tenemos un papel destacado.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


