




In addition to the classic skin manifestations of NF1 (café-au-lait spots, freckling, and neurofibromas), the presence of other manifestations such as nevus anemicus and juvenile xanthogranuloma may be of high predictive value in patients with an uncertain diagnosis. Localized or mosaic NF1, also known as segmental neurofibromatosis, is caused by a postzygotic mutation responsible for the presence of the typical disease manifestations limited to a body segment. Although no follow-up protocols are available for these patients with mosaic NF1, the risk of complications is much lower than in the generalized forms of the disease. Malignant tumors are, probably, the most feared complication of NF1. In decreasing order, the most frequent neoplasms are optic nerve glioma, malignant peripheral nerve sheath tumor, gastrointestinal stromal tumors, and pheochromocytomas. Most malignant peripheral nerve sheath tumors are the result of malignant transformation of plexiform neurofibromas and early diagnosis is a decisive factor in the prognosis. In conclusion, NF1 is a multisystemic disease that requires multidisciplinary follow-up with dermatologists playing an important role.
Además de las manifestaciones dermatológicas diagnósticas de la NF1 (MCCL, efélides y neurofibromas), existen otras alteraciones cutáneas como los nevus anémicos y los xantogranulomas juveniles, cuya presencia parece ser de gran valor predictivo en los pacientes sin diagnóstico de certeza. La NF1 mosaica localizada, clásicamente denominada neurofibromatosis segmentaria, se produce a causa de una mutación poszigótica que determina que la presencia de las manifestaciones típicas de la enfermedad se limite a un segmento corporal. Aunque no existen protocolos de seguimiento de estos pacientes, el riesgo de complicaciones es mucho menor que en las formas generalizadas de la enfermedad. Los tumores malignos son, probablemente, la complicación más temida de la NF1. En orden decreciente, las neoplasias más frecuentes son los gliomas del nervio óptico, el tumor maligno de vaina nerviosa periférica (TMVNP), los tumores estromales gastrointestinales y los feocromocitomas. La mayoría de los TMVNP son el resultado de la transformación maligna de los neurofibromas plexiformes y su diagnóstico precoz es determinante para el pronóstico. En conclusión, la NF1 es una enfermedad multisistémica que requiere seguimiento multidisciplinar, pero en cuyo diagnóstico y seguimiento los dermatólogos tenemos un papel destacado.
In addition to the classic skin manifestations of NF1 (café-au-lait spots [CaLS], freckling, and neurofibromas), the presence of other common manifestations (Table 1) may be of high predictive value in patients with typical CaLS but without definitive diagnosis. In addition, the involvement of neurofibromin in the RAS-MAPK pathway may interfere with cell proliferation and differentiation and, therefore, increase the predisposition to development of malignant tumors in these patients.
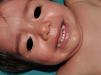
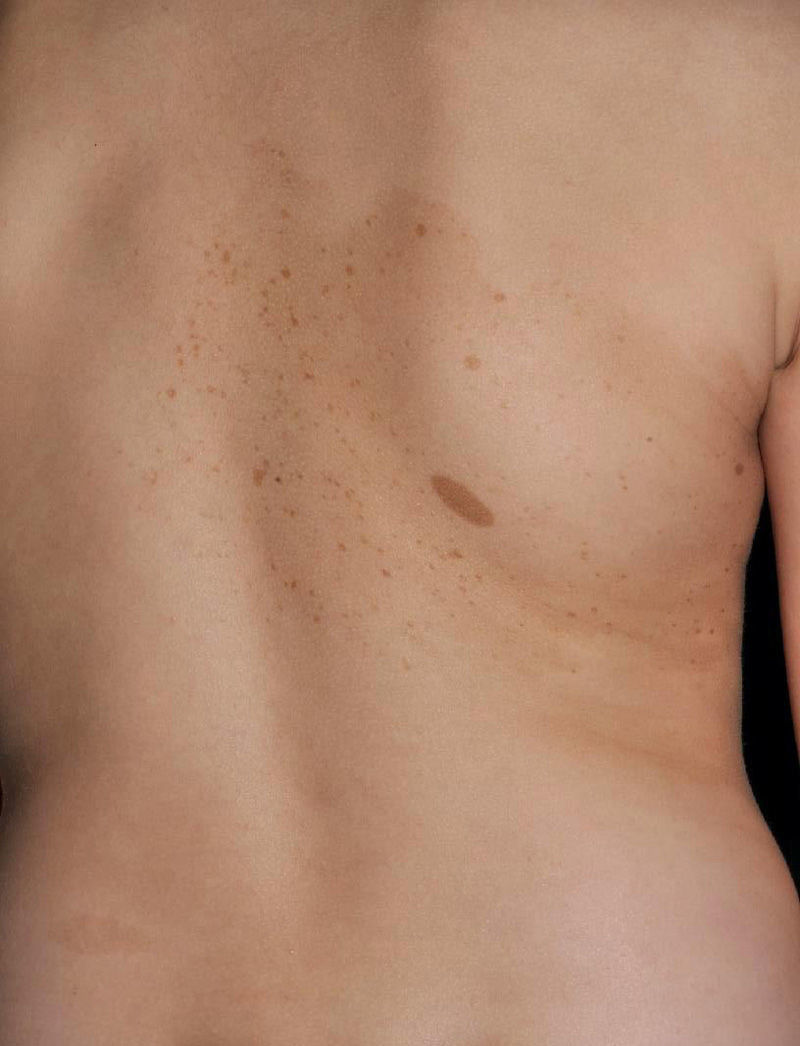
Other Common Skin Manifestations in Neurofibromatosis Type 1Nevus AnemicusThe association between NF1 and nevus anemicus (NA) was suggested by Naegeli in 1915,1 but there are hardly any literature reports on the topic until 2013.2–5 NAs are pale irregularly shaped macules whose size varies between a few millimeters and several centimeters; clinically they are not readily detected, and become apparent after mild rubbing of the area (Fig. 1). Lesions can be single or multiple and are located on any area of the body, although they are more frequent on the presternal region.3,5 Although the published series reflect a high prevalence in children, the exact time of onset of the lesions is not known, as parents often have not noticed the presence of NAs until they are detected by a dermatologist in the clinic. In our series, patients with NAs were present in at least 50% of patients with confirmed NF1.5 Possibly, the difference in prevalence in the different studies, which ranges from 8.8% to 51%,3 is a result of the retrospective or prospective study design as these lesions are difficult to diagnose unless sought systematically. To detect such lesions, we routinely ask the parents if they have seen any transient whitish lesions during bathing, fever, crying, or exercise. We then rub the entire presternal area in babies and young children to highlight the lesions. NAs appear to be due to the absence of focal response to the action of catecholamines, and histologically they are indistinguishable from normal skin.
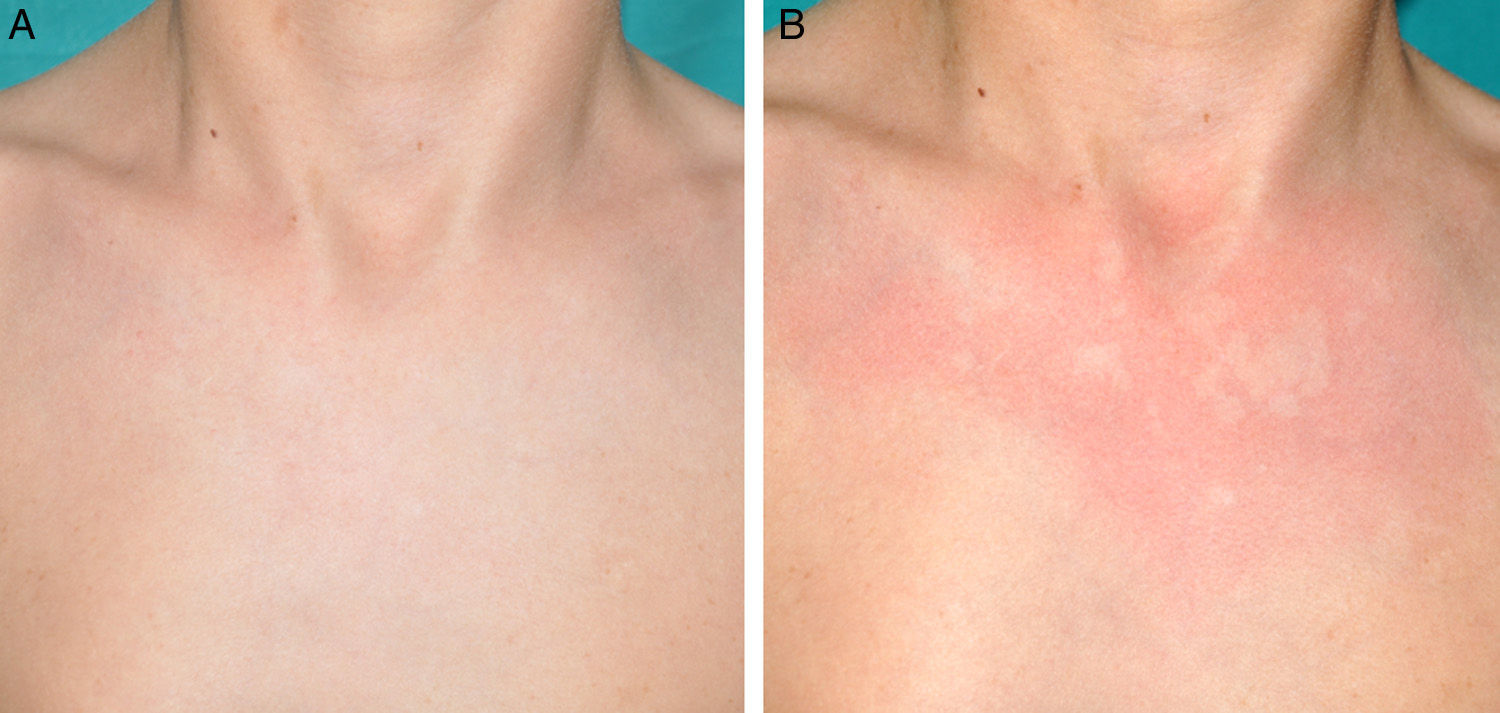
Although NAs can appear in healthy individuals, in particular, in proximity to capillary malformations, an association of these lesions and CaLS with other genodermatoses or segmental neurofibromatosis has not been reported. In line with other authors,2–4 we believe that NAs are a distinctive finding in NF1 and that the presence of these lesions is of important predictive value in NF1.5
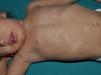
Juvenile XanthogranulomasJuvenile xanthogranulomas (JXGs) are the most common form of Langerhans cell histiocytosis, and a relatively frequent finding in NF1 (Fig. 2).6 The lesions disappear spontaneously after a few years, and it is rare to find them in late childhood and early adulthood.7 Their estimated prevalence in patients with NF1 ranges from 0.7% in adults8 to 37.5% in children,7 and these lesions are particularly common in children aged less than 9 years.4,7 In our series, the prevalence of JXGs in pediatric patients with definite diagnosis of NF1 was 6.2%,5 a figure that approximates the 8.5% reported by other authors.3 In any case, it cannot be denied that JXG is a significant finding in patients with NF1, and it can be of particular importance in cases with no definitive diagnosis. A study found that 8 out of 10 patients with NF1 who only had CaLS at the first visit and who on physical examination had JXGs or NA, subsequently developed the disease,4 and another study found that NF1 was subsequently confirmed in children with JXGs and CaLS.7 Therefore, considering that JXG is the most frequent tumor in NF1, it is probable that the coexistence of CaLS and JXGs in small children should not be considered a casual finding but rather one highly indicative of the disease. Their presence has also been associated with a greater risk of childhood leukemia in patients with NF1, in particular juvenile myelomonocytic leukemia (JMML).9 However, this association is not supported by the findings of other studies, and has been questioned by some authors,10–12 in particular in the dermatology literature. Although there are several case reports of such an association,13–16 a greater incidence of JMML has not been observed in larger series of patients with NF1 who presented with JXGs, including our series.4,5,7 Therefore, the suspicion of JMML in a patient with NF1 who develops JXGs should be directed by clinical symptoms and physical exploration, and not by the mere finding itself.11
Glomus Tumors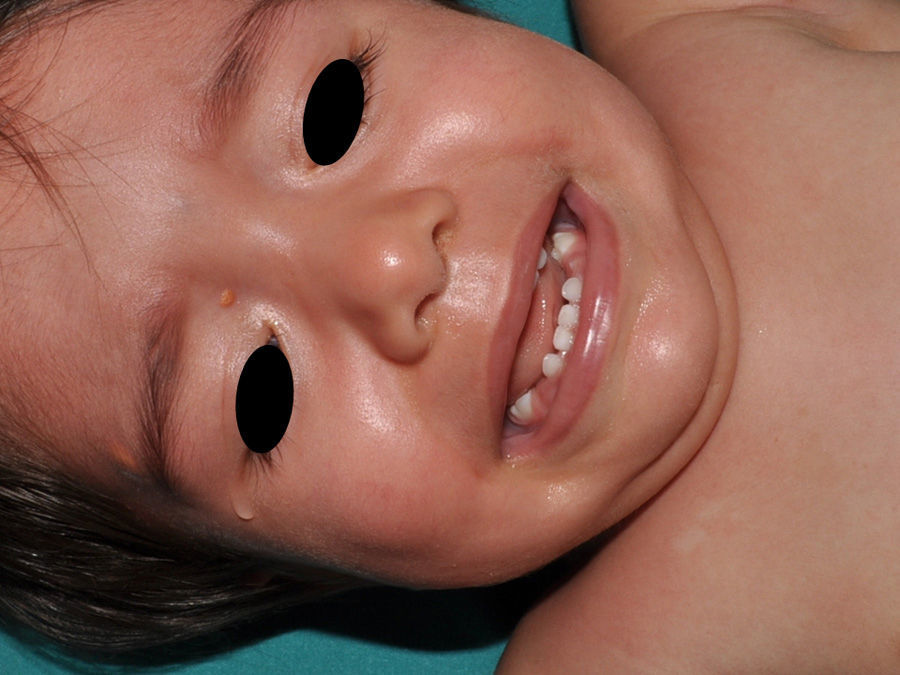
The association of NF1 with glomus tumors was suspected since 1938,17 but it was not until 2009 when this association was definitively confirmed when NF1 mutations on both alleles was demonstrated in glomus cells.18 Glomus tumors are benign vascular lesions that originate in the glomus body, a neuro-myo-arterial structure specialized in the regulation of vascular flow. The lesions are usually located in acral regions, particularly below the nails, and they are characterized by paroxysmal pain on applying pressure and with temperature changes. In patients with NF1, these lesions are usually multiple and recurring.19 They are rare in children, with an estimated prevalence of 5% in adult patients with NF1.20 Although they can appear in healthy individuals, around 30% of patients with glomus tumors have NF1.19,20 From the histological point of view, they are characterized by the presence of fine-walled vessels surrounded by uniform cells with rounded and dark nuclei. Unlike in sporadic cases, these cells do not express neurofibromin in individuals with NF1.19
Other Common Skin Findings in Neurofibromatosis Type 1Pruritus, generalized hyperpigmentation, presence of hypochromic macules, and soft skin are findings that can be readily detected in patients with NF1.
Pruritus affects 20% of patients with NF1 and considerably impacts their quality of life.21 The itching has been attributed to increased mastocytes in the skin, but there do not appear to be any differences in plasma levels of histamine in patients with itching compared to the general population.22 Many patients do, however, have pruritus localized in the neurofibromas, where elevated mastocytes have been detected; administration of ketotifen can suppress the itching.23 We should bear in mind that localized itching can be a warning for the development of medullary tumors or tumors of the central nervous system,24,25 and so it is a relevant symptom in patients with NF1.
Patients with NF1 have diffuse generalized hyperpigmentation that becomes evident on comparing the tone of their skin with that of unaffected family members, and this explains the underlying hyperpigmentation often seen in segmental forms of NF1. Recently, in vitro studies have shown that melanocytes with a mutation in the NF1 gene reproduce this hyperpigmentation, which is attributed to overexpression of microphthalmia-associated transcription factor, tyrosinase, and dopachrome tautomerase, all agents involved in melanogenesis.26
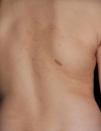
Just as CaLS are observed in tuberous sclerosis, in children with NF1, hypopigmented macules can be found relatively frequently (Fig. 3),27 but to date, there are no studies that have characterized their prevalence, number, and morphology. Finally, skin softness in patients with NF1 is a subjective perception that is also highlighted by some experts in the disease.27
Segmental or Mosaic Localized Neurofibromatosis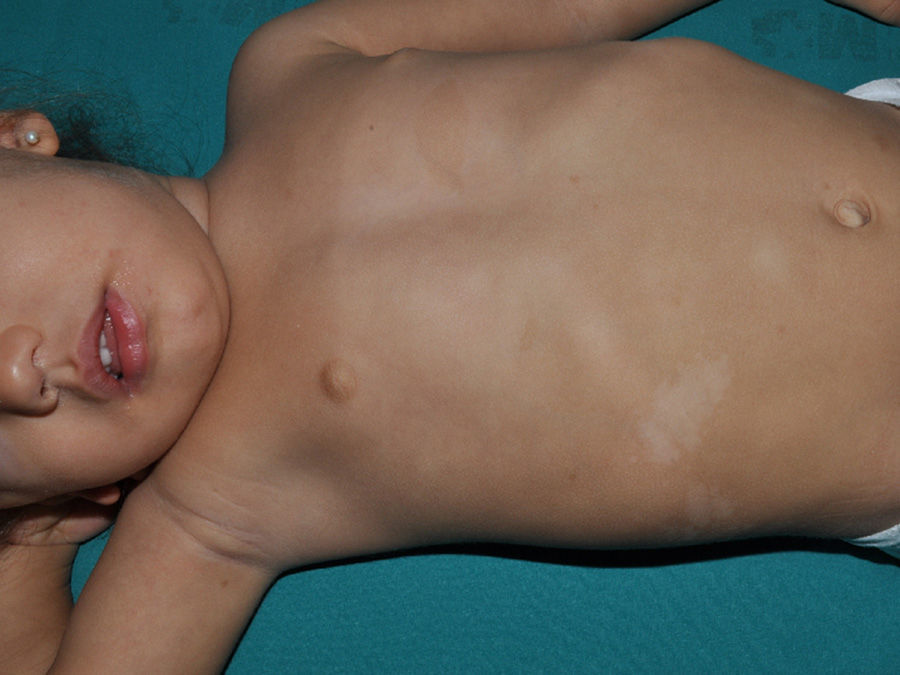
Currently, segmental neurofibromatosis tends to be denoted mosaic localized neurofibromatosis (MLNF). Characteristically, typical lesions of NF1 are limited to one or several body segments. Four different types can be distinguished according to the clinical findings: 1) MLNF with exclusively pigmentary cutaneous changes (CaLS and freckling); 2) MLNF with neurofibromas alone; 3) MLNF with cutaneous pigmentary changes and neurofibromas; and 4) MLNF with exclusively plexiform neurofibromas.28 A minimal number of CaLS is not required and the freckling does not have to be localized to skin folds to enable diagnosis of LMNF. Likewise, there is no minimum number of 2 cutaneous neurofibromas in the region of skin with typical pigmentary changes. The prevalence is estimated to be between 1:30 000 and 1:40 0000 healthy individuals,8 but it is probably more common, as it often goes unnoticed by physicians and parents, who consider it a birthmark. The development of lesions follows the same time course as that of the systemic form of the disease. Therefore, most patients will only have pigmentary lesions during childhood and develop neurofibromas from puberty onwards.28,29 In the forms with exclusively pigmentary changes, tenuous underlying hyperpigmentation is observed that delimits the affected segment which, although well defined, can slightly exceed the medial line (Fig. 4). Some patients present with CaLS outside the apparently affected segment, particularly when the segment involves the scapular or pelvic region. In these patients, CaLS may be present along the involved limb. Other cutaneous findings, such as NAs or JXGs have not been described, and systemic complications that are often associated with NF1 appear to be less frequent in MLNF.30 MLNF is usually asymptomatic but it can cause itching,31 or pain resulting from nerve root compression or malignant transformation.32,33
It is not uncommon for MLNF to be confused with other lesions such as nevus spilus, pigmentary mosaicism, or, very occasionally, segmental lentiginous pigmentation.34 These pigmentation disorders are very difficult to distinguish clinically from MLNF, and differentiation can only be achieved by molecular analysis of the tissue. Patients with mosaic forms of NF1 should not be diagnosed with generalized NF1, even when they meet the diagnostic criteria for number and size of CaLS in the affected area. Nevertheless, patients should be aware of a small risk of transmitting generalized disease to their descendents.28,30 It is important to highlight that gonadal mosaicism is independent of the site of the affected cutaneous area, which is not necessarily localized on or near the gonadal region. Furthermore, there are no conclusive data on the risk of germline mosaicism. A recent systematic review did find that 4 of 157 cases published (2.5%) transmitted a generalized form of the disease,30 but the authors themselves pointed out the possibility of publication bias. In addition, we do not have any data on the total number of descendants of patients with MLNF reported or the percentage of offspring affected. This information is essential for reliably calculating the risk of transmission of the disease.
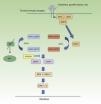
Neurofibromatosis Type 1 and CancerMalignant tumors are, probably, the most feared complication of NF1. The signaling pathway mediated by the MAP-kinases ERK1 and ERK2 plays an essential role in the control of cell proliferation, differentiation, and survival in physiological conditions, and so failures associated with regulation of this pathway significantly contribute to cell transformation, and these are undoubtedly involved in tumor progression (Fig. 5). In decreasing order, the most frequent neoplasms in NF1 are optic nerve glioma (ONG) (15%-20%), malignant peripheral nerve sheath tumor (MPNST) (8%-13%), gastrointestinal stromal tumors (4%-25%), and pheochromocytomas (0.1%-5%).35 In addition, patients with NF1 are at 7 to 5 times greater risk than the general population of developing leukemia, brain tumors, and breast cancer. Finally, although the relationship between NF1 and melanoma is not clear, recent findings have implicated neurofibromin in the genesis of this tumor. For reasons of space, we will only discuss tumors of neural origin and the relationship between neurofibromin and melanoma.
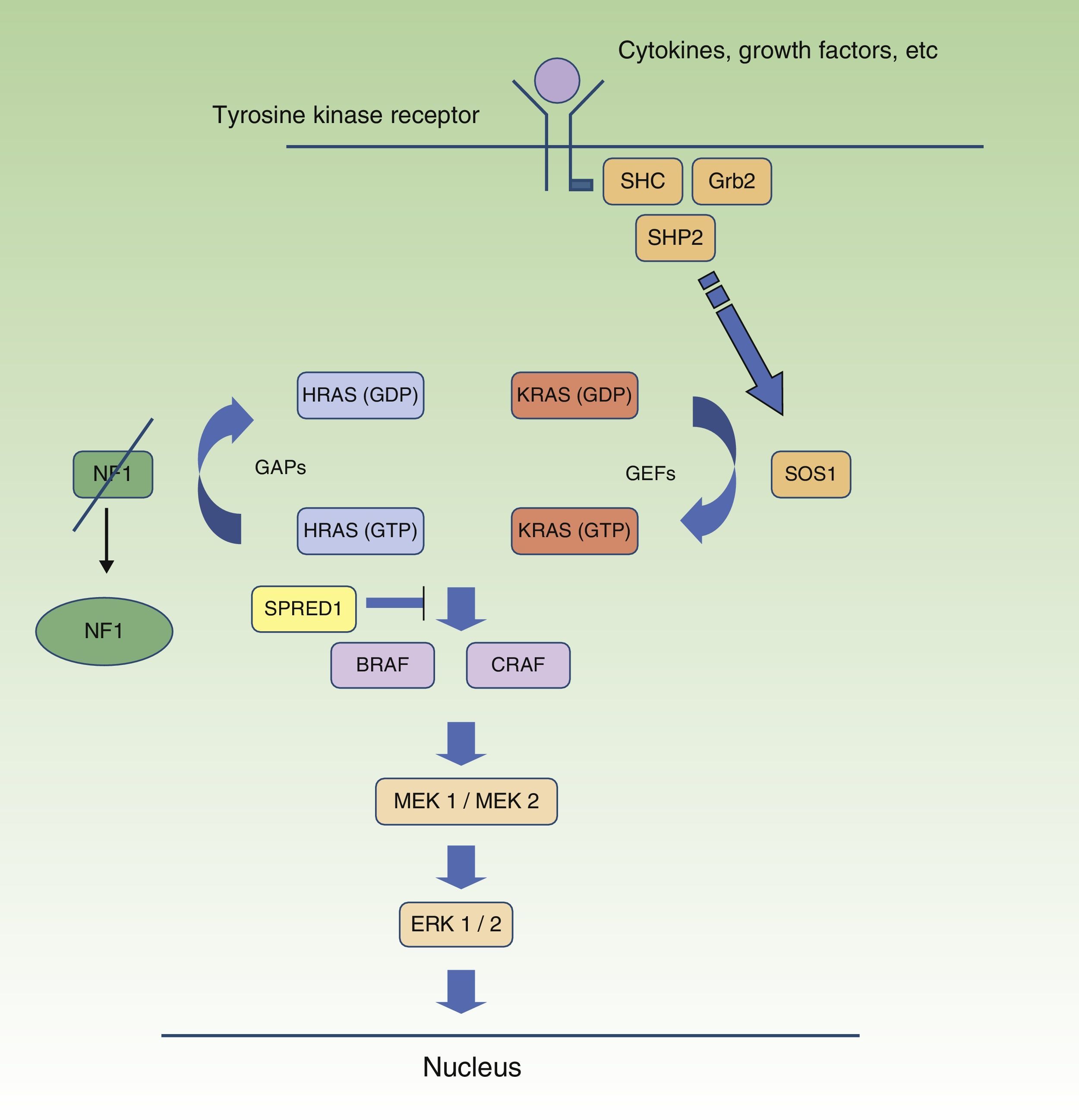
RAS MAPK metabolic pathway. After stimulation of the cell receptors, intracellular proteins are activated (SHC,GRB2, and SHP2), and these recruit intracytoplasmic SOS1, activating the RAS proteins. Activation of the RAS proteins is accompanied by activation of RAF (BRAF, RAF1), MEK1A1/MEK1A2 and, finally, ERK/ERK2, which are the last effectors of the RAS/MAPK pathway and responsible for maintaining the cell life cycle.
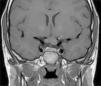
ONGs account for 85% of low-grade gliomas (pilocytic astrocytomas) in patients with NF1, and affect between 15% and 30% of children aged between 3 and 5 years.36,37 They are a diagnostic criterion for the disease. Most cases are asymptomatic, but they can cause loss of vision in up to 12% of patients37 or provoke early onset of puberty.35 In view of the high incidence of ONGs, close ophthalmological follow-up is advisable in children with NF1, whether this is clinical or radiological, as clinical detection by physical examination can be difficult and some neuropediatricians prefer conducting complementary tests (Fig. 6).37 The remaining 15% of low-grade gliomas also occur more frequently in the first 10 years of life, and can appear in any part of the brain. They are manifest clinically as headaches, unstable gait, or lethargy.35 By contrast, high-grade gliomas (glioblastoma multiforme) are more often seen in young adults. The risk of such tumors is 5-fold greater in patients with NF1 than in the general population and prognosis is poor.35,38 Treatment for ONG in patients with NF1 is usually conservative, unless a decrease in visual acuity or progression in the size of the tumor in imaging tests is observed. If necessary, first-line treatment is chemotherapy with vincristine and carboplatin. Radiotherapy is not recommended given the high risk of secondary tumors. Surgery is reserved for large orbital tumors in which the patient has no useful vision, when the cornea is exposed, or if there is proptosis.39
Malignant Peripheral Nerve Sheaf Tumor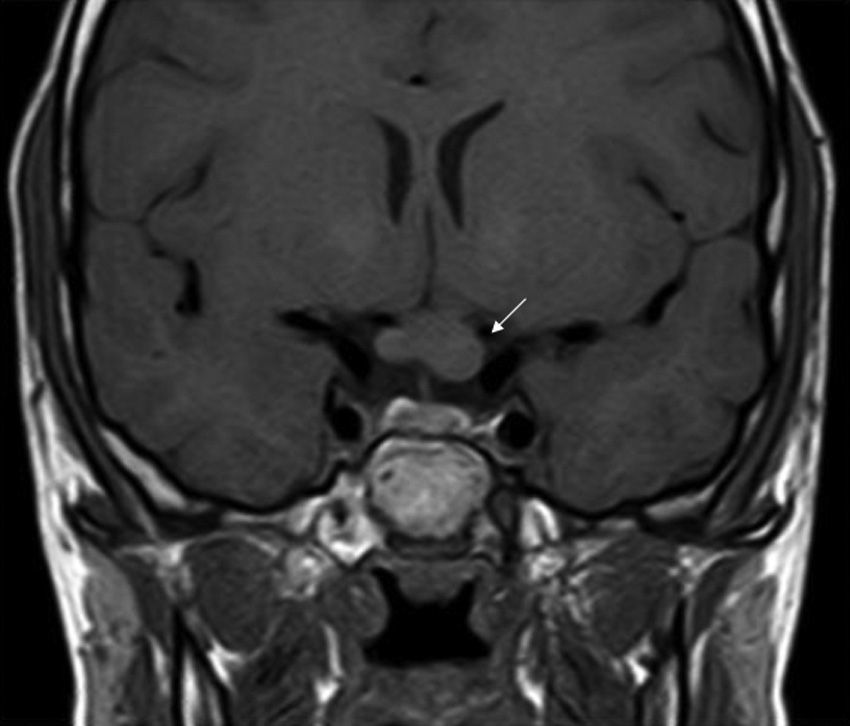
MPNST (or neurofibrosarcoma) is a type of sarcoma that is thought to be derived from Schwann cells. It accounts for 3% to 10% of all sarcomas and is almost always associated with NF1. The approximate prevalence in patients with NF1 is 0.1% and the cumulative life-long risk is 8% to 13%.35 In NF1, these tumors present in less than 5% of adult patients40,41 and in less than 2% of children.36 The sites at greatest risk are areas with preexisting plexiform neurofibroma, when there is complete deletion of the gene or a history of prior radiotherapy. Factors predictive of the presence of internal (or deep) plexiform neurofibromas have been discussed in another section, but although the majority of MPNST result from malignant transformation of plexiform neurofibromas, they can also appear on subcutaneous neurofibromas and even where there are no prior lesions.36,40,42 The molecular mechanisms of malignant transformation of neurofibromas are not well known, but mutation of the NF1 gene on both alleles in tumors, along with other epigenetic mechanisms, may lead to mutation of other important genes such as p53 and InK4A.41 Warning symptoms include sudden increase in size, change in texture (increased consistency), intense or unremitting pain, and neurological symptoms other than those usually present.40 In these cases, a radiological assessment and biopsy (with surgical resection if appropriate) should be performed immediately as a delay in diagnosis (in direct relation to the tumor size) is directly correlated to prognosis.43 It should be remembered that magnetic resonance imaging offers data on the extent and site, but not on the biological behavior of the tumor. For such information, positron emission tomography with 18-fluorodesoxyglucose is necessary. This imaging technique will also provide information on potential metastases. Fine needle aspiration biopsy is not necessary as the histological sample should provide enough tissue sample for analysis. Radical surgery is the only curative therapeutic procedure, but it is often impossible or too disfiguring. Use of coadjuvant chemotherapy is controversial and not standard practice, except the use of anthralin in patients with metastatic tumors.44,45 Currently, MPNST has poor prognosis for survival.35
MelanomasIt is not clear whether there is a true association between NF1 and melanoma or whether the published cases, which are usually sporadic, are merely anecdotic. In the largest series, which included 11 patients with NF1 and cutaneous melanoma, melanoma was found to occur more frequently in women (in a proportion of 1:10), and young people (median age, 33 years). The mean Breslow depth was 3.2mm, that is, considerably greater than in other series of patients with melanoma without NF1,46 perhaps because other pigmented lesions make detection more difficult. Despite the uncertainty of the association, the relationship between melanoma and the RAS pathways is irrefutable, as not only do 50% of melanomas have BRAF mutations (particularly V600E) and 15% to 20% of these have NRAS mutations (particularly in codon 61) but also many melanomas have NF1 mutations, whether associated or not with the aforementioned mutations.47 The NF1 gene is therefore the third most affected gene in melanoma. NF1 mutations are particularly common in desmoplastic melanomas, where they are detected in 93% of cases, compared with 20% in nondesmoplastic melanomas.48 However, the predictive value for response to treatment with MEK inhibitors of the presence of these mutations is subject to debate.49,50 In addition to melanoma, mutations in the neurofibromin gene have been detected sporadically in other cancers, such as brain tumors, breast cancer, ovarian cancer, and leukemias, and this is a promising line of research.51
Finally, it is important to note that the rate of mortality due to cancer in individuals with NF1 is somewhat higher in individuals under 50 years of age, with this increased rate driven mainly by breast cancer and JMML, but the rates of cancer compared with the general population even out above this age.52 Likewise, although the mean life expectancy is somewhat lower in patients with NF1 than in healthy individuals, the majority of NF1 patients live past 70 years of age.
Diagnosis of Neurofibromatosis Type 1Currently, diagnosis of NF1 is still mainly based on clinical observations.53 However, the timing of development of diagnostic criteria hinders diagnosis in small children and, in fact, only 46% of patients with no family history of the disease are diagnosed before the age of 2 years.54 In recent years, several authors have proposed the inclusion of other cutaneous findings such as NAs and xanthogranulomas in the diagnostic criteria to facilitate diagnosis in this age group,2–5,7,27 and some authors go as far as to propose a revision and update of the classical clinical criteria.27,55
Genetic diagnosis of NF1 is not generally available. The large size of the NF1 gene, the presence of homologous pseudogenes in pericentric regions of other chromosomes, the limited genotype-phenotype correlation, and the absence of hotspots make study of mutations laborious and costly. The combination of molecular techniques increases the sensitivity of detection of genetic mutations. Specifically, the combination of RNA analysis (denaturing high performance liquid chromatography) and multiplex ligation-dependent probe amplification, validated by Spanish researchers, has a sensitivity of 95% for detection of mutations in the NF1 gene and provides very reliable results.56 To date, more than 1000 mutations have been reported throughout the entire NF1 gene; the majority of NF1 mutations occur due to point changes in the gene coding sequence, while gene deletion occurs only in a minority of cases. Although the penetration of the disease is almost 100% in adults, there is good genotype-phenotype correlation in only a very few cases, and such correlation is not usually seen even in the same family or identical twins.57–60 The exceptions are cases of complete microdeletion of the NF1 gene of 1.4 Mb (patients with such mutations have a severe form of the disease, with numerous and early-onset neurofibromas, cognitive impairment, dysmorphic features, and a tendency to develop malignancies),58,61 deletions of 3 base pairs in exon 17 affecting a single amino acid, p.Met992del (cases present CaLS and freckling without neurofibromas),60 and mutations in codon 1809 of exon 29, which presents with pulmonary stenosis and short stature (phenotypic characteristics of Noonan syndrome).59
Problems with the genetic study have led some authors to consider routine genetic studies unnecessary in small children who still do not meet the criteria for the disease, as: 1) 95% of children will meet the clinical criteria at the age of 8 years; 2) a negative result in the genetic test, in absence of a known familial mutation, does not rule out diagnosis; and 3) the absence of correlation between genotype and phenotype does not allow a prognosis to be made or change the approach to follow-up.62,63 Set against this opinion is the day-to-day reality of physicians who attend children with NF1: worried parents (who are perfectly informed about the risks and worst aspects of the disease) who want to know for sure whether their child has the disease. It is likely that advances in molecular diagnostic techniques will decreases costs and genetic studies will become widespread.
In conclusion, the presence of cutaneous findings such as NAs and JXGs seem to be of high predictive value in patients with CaLS but with no definite diagnosis of NF1. Patients with NF1 have a predisposition to cancer, and early diagnosis of malignant tumors is the determining factor in prognosis. NF1 is a multisystemic disease that requires multidisciplinary follow-up with dermatologists playing an important role.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Hernández-Martín A, Duat-Rodríguez A. Neurofibromatosis tipo 1: más que manchas café con leche, efélides y neurofibromas. Parte II. Actualización sobre otras manifestaciones cutáneas características de la enfermedad. NF1 y cáncer. Actas Dermosifiliogr. 2016;107:465–473.


