







Neurofibromatosis type 1 (NF1) is the most common neurocutaneous syndrome and probably the one best known to dermatologists. Although the genetic locus of NF1 was identified on chromosome 17 in 1987, diagnosis of the disease is still based primarily on clinical observations. The 7 diagnostic criteria of the National Institutes of Health, which were established in 1988, include 3 skin manifestations (café-au-lait spots, freckling on flexural areas, and cutaneous neurofibromas). The age at which these diagnostic lesions appear is variable: onset can be late in some patients while others never develop certain symptoms. Definitive diagnosis may therefore be delayed by years. Although the appearance of the characteristic café-au-lait spots and freckling in the early years of childhood are very suggestive of the disease, these signs are not pathognomonic and, in isolation, do not constitute sufficient evidence to establish a definitive diagnosis. Thus, other diagnoses should be considered in patients whose only symptoms are café-au-lait spots and freckling. By contrast, the presence of multiple cutaneous neurofibromas or at least 1 plexiform neurofibroma is a very specific indication of NF1. Identification of the different types of neurofibroma allows us to confirm the diagnosis and initiate appropriate management.
La neurofibromatosis tipo 1 (NF1) es el síndrome neurocutáneo más frecuente y probablemente el mejor conocido por los dermatólogos. Aunque desde 1987 se sabe que el locus genético de la NF1 se localiza en el cromosoma 17, en la actualidad el diagnóstico de la NF1 sigue siendo fundamentalmente clínico. Los criterios diagnósticos del National Institute of Health están vigentes desde el año 1988 e incluyen 7 criterios diagnósticos, de los cuales tres se manifiestan en la piel: las manchas café con leche [MCCL], las efélides flexurales, y neurofibromas cutáneos. La edad de aparición de los distintos criterios diagnósticos es variable, y algunos pacientes los manifiestan tardíamente o incluso no los desarrollan nunca, por lo que el diagnóstico de certeza puede demorarse durante años. Las MCCL y las efélides aparecen en los primeros años de la vida y son muy sugerentes de la enfermedad, pero no son patognomónicas y resultan insuficientes para realizar el diagnóstico de certeza. Así, cuando los pacientes presentan exclusivamente MCCL y efélides es imprescindible considerar otros diagnósticos. Por el contrario, la existencia de múltiples neurofibromas cutáneos, o al menos un neurofibroma plexiforme, son muy específicos de la NF1. La identificación de las distintas formas de neurofibromas nos permite confirmar el diagnóstico de la enfermedad y hacer un seguimiento adecuado de los mismos.
Neurofibromatosis type 1 (NF1) (Online Mendelian Inheritance in Man [OMIM] #162200) is the most frequently occurring neurocutaneous syndrome. It has an estimated prevalence of 1:2500-1:3000 among live newborn babies,1,2 and it is the most well-known neurocutaneous disease among dermatologists, who are usually the first specialists to suspect and/or confirm its diagnosis. Although the genetic locus of NF1 was identified on chromosome 17 in 1987,3 diagnosis is still mainly based on clinical observations and the diagnostic criteria of the National Institute of Health (NIH), dating from 1988 and still in force today (Table 1).4 NF1 can affect almost all organs and body systems, leading to learning disorders and ophthalmological, neurological, orthopedic and cardiovascular problems, as well as tumors. In this review, we will focus on the dermatological manifestations and the relationship between NF1 and cancer.
Clinical Diagnostic Criteria for Neurofibromatosis Type 1 According to National Institutes of Health1 in Force Since 1988 (At Least 2 Criteria Are Required).
| Six or more café au lait spots over 5mm in diameter in prepubertal individuals and over 15mm in postpubertal individuals |
| Freckling in the axillary or inguinal regions |
| Two or more neurofibromas of any type or 1 plexiform neurofibroma |
| Optic nerve glioma |
| Two or more Lisch nodules |
| An osseous lesion suggestive of neurofibromatosis type 1 such as sphenoid dysplasia or thinning of long bone cortex with or without pseudoarthrosis |
| A first-degree relative with neurofibromatosis type 1 by the above criteria |
The NF1 gene is located in the pericentromeric region of chromosome 17 (17q11.2). It is formed of 60 exons of between 100 and 200 base pairs that extend across 282kb of DNA. It encodes a protein known as neurofibromin, which is expressed in Schwann cells, melanocytes, leukocytes, the adrenal gland, and tissues such as the central nervous system.3 Neurofibromin contains a central segment denoted NF1-GRD, which has an inhibitory effect on the mitogen activated protein kinase enzymatic pathway (RAS/MAPK).5 The RAS pathway switches on signal transduction from the outside to the inside of the cells, thereby activating a cascade of proteins implicated in developmental processes (proliferation, cell differentiation, organogenesis, synaptic plasticity, growth, apoptosis, and cell ageing). The involvement of neurofibromin in the RAS-MAPK pathway not only increases predisposition to tumors, but is also associated with presence of phenotypic anomalies and learning and memory impairments, a characteristic shared with other RASopathies.6
NF1 is of autosomal dominant transmission, but half the cases are sporadic or de novo, and the patients have no family history of the disease. In addition, there are mosaic forms of the disease in which a late somatic mutation may give rise to mosaic neurofibromatosis with manifestations limited to one or several body segments (localized mosaicism or segmental neurofibromatosis), whereas early mutations give rise to a condition clinically indistinguishable from the generalized forms (generalized mosaicism).7 Type 2 mosaicism (in which a more affected segment is superimposed on generalized disease) is much less common in NF1 and to date has only been shown in segments already affected by neurofibromas.8
Skin Manifestations Diagnostic of Neurofibromatosis Type 1Cutaneous findings, which are readily apparent on visual inspection, are usually the first sign of the disease. Three of these, cafe-au-lait spots (CaLS), freckles on flexural areas, and neurofibromas are particularly relevant because they comprise 3 of the 7 clinical diagnostic criteria of the NIH.
Café-au-Lait SpotsCaLS are, probably, the most important clinical criterion in children, because they are present in almost all patients with NF1 and because they are usually the first visible sign of the disease.9,10 CaLS are usually present at the time of birth or they become evident shortly afterwards. In our experience, 90% of patients already have a sufficient number of CaLS within the first year of life to meet the diagnostic criterion.11 Their size increases in proportion with the growth of the child, and they take on a darker tone during childhood,12 before lightening again during adulthood.13 Although they can be found anywhere on the body, they do not usually appear on the face.
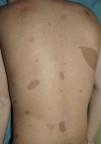
From the morphological point of view, typical CaLS have a variable color, homogenous tone, and a smooth, regular border (Fig. 1). They also vary in size considerably, from a few millimeters to several centimeters, but to be considered a criterion for NF1, there need to be at least 6 lesions equal or greater than 5mm in diameter at prepubertal ages or greater than 15mm after puberty. Exceptionally, some patients with definite diagnosis of NF1 do not have CaLS, have an insufficient number of them, or the lesions have an atypical morphology.11,14 However, most patients with more than 6 CaLS without other markers of NF1 will end up developing NF1, usually before they are 5 years old, and almost always before they are 8 years old.14,15 The more typical the morphology of CaLS, the greater the probability that NF1 is confirmed, and there is no association between the number of CaLS and the severity of NF1.14 Histologically, CaLS have increased melanin in melanocytes and basal keratinocytes, but no melanocyte proliferation.16 The presence of macromelanosomes has also been observed, along with a greater concentration of melanin in CaLS of patients with NF1 than in sporadic CaLS.17,18 From the molecular point of view, it has been demonstrated that melanocytes of CaLS have a second NF1 mutation in the gene on the healthy allele.19
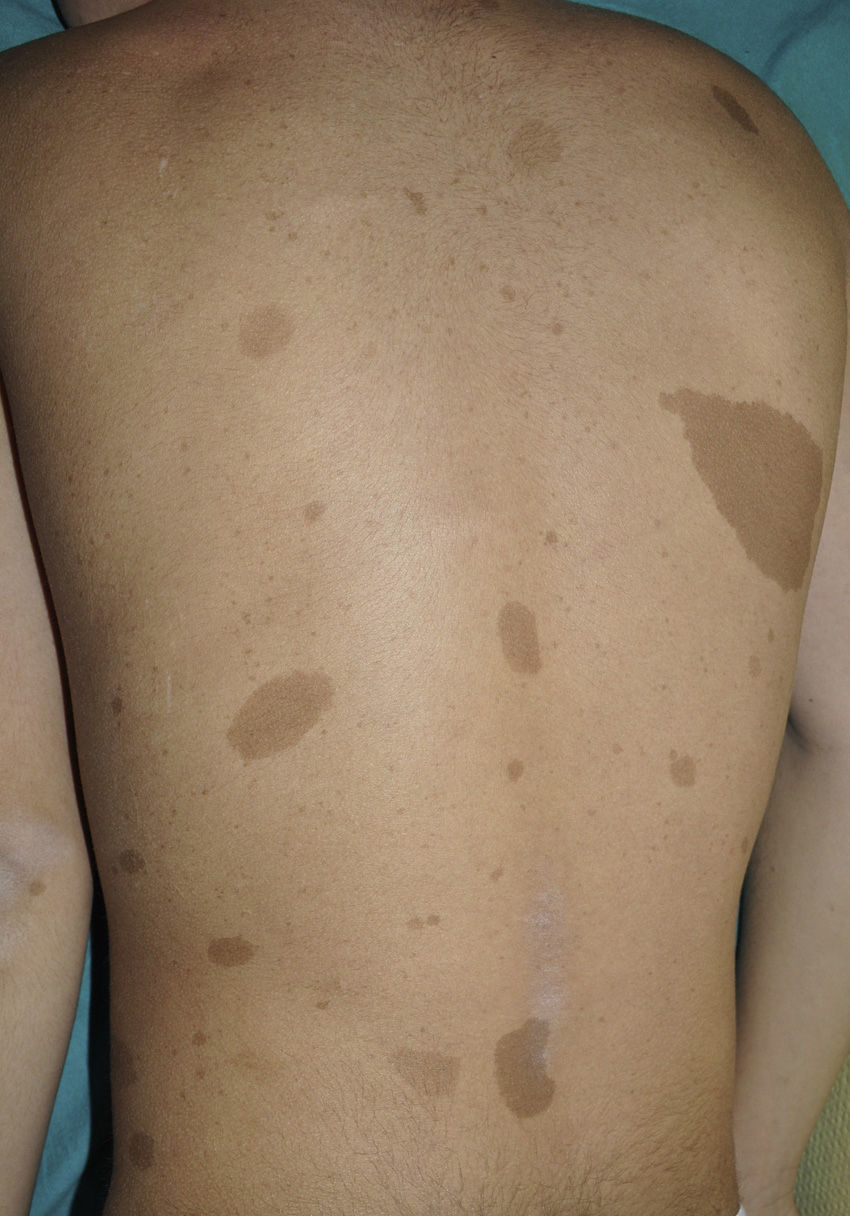
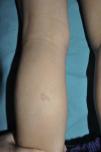
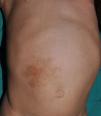
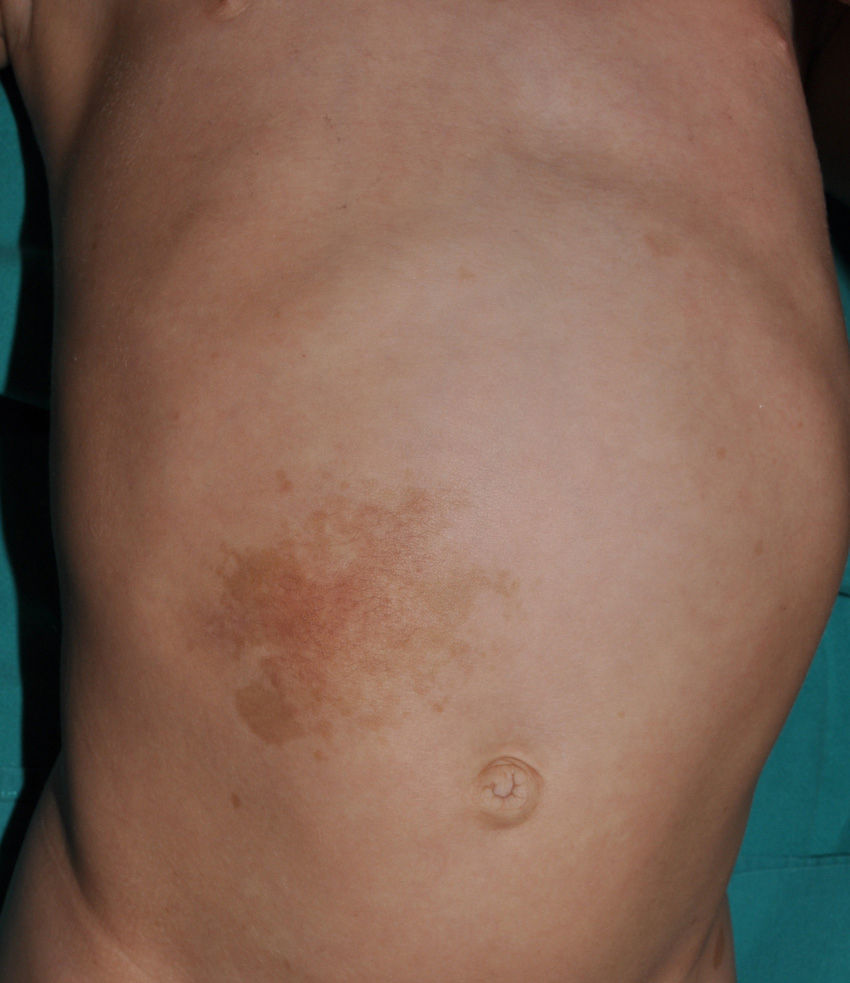
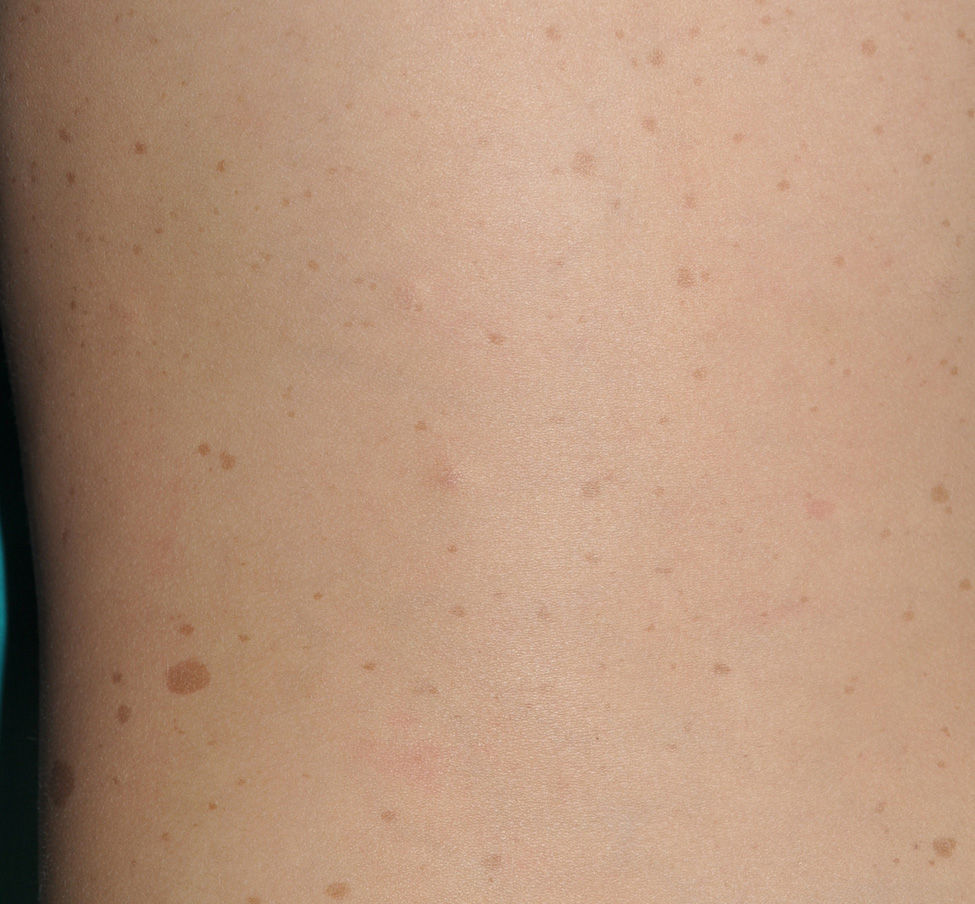
Small children with CaLS and NF1 are often referred to our clinics. However, although CaLS are very suggestive of NF1, they are not pathognomonic. It is calculated that up to 20% of children have isolated CaLS, 4% have 2, and fewer than 1% of the healthy population have more than more than 3.20,21 The physical examination and medical history should be particularly rigorous in those individuals with 3 lesions. The first thing that we should assess is whether the lesions are actually CaLS. That is, we should differentiate these lesions from others such as hyperchromic nevi (which can be multiple but they have a much more irregular border) (Fig. 2), pigmentary mosaicisms, congenital melanocytic nevi, nevus spilus, macular urticaria pigmentosa, and postinflammatory hyperpigmentation. Occasionally, in some patients with NF1, we can observe extensive hyperpigmented macules with irregular borders, with or without hypertrichosis, which are confused with giant CaLS and which may correspond to superficial plexiform neurofibromas (Fig. 3). After assessing the number and size of CaLS, other clinical manifestations should be assessed to confirm diagnosis of NF1. This can be difficult in very young children, as although the NIH clinical criteria are very specific and sensitive in children aged over 8 years, no more than 45% of children less than 1 year of age meet them.15 Therefore, it is essential to perform complementary examinations and collaborate with other specialists, particularly neuropediatricians and ophthalmologists.
Several diseases may be associated with CaLS (Table 2). In practice, RASopathies, and Legius syndrome in particular, are the most important conditions to include in the differential diagnosis. Legius syndrome (OMIM #611431) or NF1-like syndrome is a RASopathy described in 2007 that is very similar to NF1.22 It is caused by a mutation in the sprouty related EVH1 domain-containing protein 1 (SPRED1) gene, which is located on chromosome 15q13.2 and is comprised of 7 exons. Mutation of this gene is associated with loss of RAF protein suppressor function. Legius syndrome is of autosomal dominant transmission and is characterized by the presence of CaLS and/or freckles, which are occasionally associated with macrocephaly, lipomas, Noonan phenotype, and/or learning difficulties.23 Often, these patients meet sufficient pigmentary criteria for diagnosis of NF1, but in Legius syndrome the presence of Lisch nodules, neurofibromas, central nervous system tumors, and NF1 gene mutations are specifically excluded. However, the probability of finding a mutation only in the SPRED1 gene in a patient with CaLS and freckles is no greater than 2%, and so many laboratories only analyze the SPRED1 gene when mutations in the NF1 gene have been ruled out. This approach detects mutations in up to 75% of patients with CaLS and freckles in flexural areas.24 In particular, deletions of a single amino acid, p.Met992del, in the NF1 gene, leads to a phenotype that is indistinguishable from Legius syndrome.25 In 20% of patients with CaLS and freckles only, there are no mutations in either of these genes.24 It seems that the probability of finding patients with Legius syndrome is greater in patients with a family history of CaLS with or without freckles.23
Differential Diagnoses of Neurofibromatosis Type 1.
| Café-au-Lait Spots |
| Legius syndrome |
| LEOPARD/Noonan syndrome with lentigines |
| Familial café-au-lait spots |
| McCune-Albright syndrome |
| Neurofibromatosis type 2 |
| Albinism |
| Ring-chromosome syndromes |
| Constitutional mismatch repair syndrome |
| Others: Noonan syndrome, cardio-facial-cutaneous syndrome, tuberous sclerosis, Bloom syndrome, ataxia-telangiectasia, Fanconi anemia, Russell-Silver syndrome, Turner syndrome, MEN I and MEN II syndromes, Johanson-Blizzard syndrome, Rubenstein-Taybi syndrome. |
| Diseases with Freckles |
| Legius syndrome |
| LEOPARD/Noonan syndrome with lentigines |
| Peutz-Jeghers syndrome |
| Cowden syndrome |
| Carney syndrome |
| Others: syndromes with photosensitivity, simple epidermolysis bullosa with mottled pigmentation |
| Diseases With Subcutaneous Tumors |
| Lipomatosis |
| Proteus syndrome and other overgrowth syndromes |
| Complex combined vascular malformations |
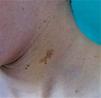
CaLS that appear in other RASopathies have not been well characterized in the literature, with the exception of LEOPARD syndrome, but in our experience, these rarely meet the criteria for NF1. Although for some time there was talk of the NF1-Noonan syndrome to describe children with the Noonan phenotype and pigmentary cutaneous manifestations fully typical of NF1, subsequently, it was shown that these patients had mutations in the NF1 gene and they actually did not have Noonan syndrome but a true form of NF1.26 Specifically, missense mutations in the NF1 gene that affect the p.Arg1809 residue have typical phenotypes of Noonan syndrome (pulmonary stenosis and short stature) and typical pigmented lesions of NF1, but absence of neurofibromas.25 CaLS of patients with LEOPARD syndrome usually have a darker tone (past literature has described these as black coffee spots instead of CaLS), often with a heterogeneous color. The borders usually tend to be more polygonal than rounded, as reflected in several images in the literature, including some of our patients (Fig. 4).27–29 Lentigines or generalized freckles, which appear progressively from 4 or 5 years of age, are much more characteristic of LEOPARD syndrome than CaLS, but they may be similar to those that present in patients with NF1. Diagnosis is hindered further by the fact that the most typical systemic manifestations of LEOPARD syndrome, such as deafness and pulmonary stenosis, may not be present,27 and so molecular diagnosis is essential in some cases to differentiate between conditions. Finally, other RASopathies such as Costello syndrome and cardio-facial-cutaneous syndrome may be associated with CaLS, but this only occurs in a minority of patients and the number of lesions is usually small.30–32
Café-au-lait spots with polygonal shapes in 2 patients with LEOPARD syndrome.
Source: Taken from Ramos-Geldres et al.27
Familial CaLS should be suspected in individuals from different generations of CaLS without any other manifestations of NF1. The condition is believed to be of autosomal dominant transmission, but the disease has still not been characterized at a molecular level. Familial CaLS may correspond to a form of Legius syndrome, although this has been ruled out in some cases.24 Mosaic forms of NF1 may exist exclusively with cutaneous involvement and NF1 mutations may not be detectable in circulating leukocytes.19 It may even be the case that the mutation resides in another gene of the RAS pathway.33,34 McCune-Albright syndrome is characterized by extensive CaLS, often with a unilateral segmental distribution and irregular borders. Polyostotic fibrous dysplasia, early puberty, and other endocrine disorders may also be present. The main diagnostic problem comes with the segmental forms of NF1 and with hyperpigmented cutaneous mosaicisms. Neurofibromatosis type 2 may be associated with CaLS in 32% to 47% of patients,35 but more typical are schwannomas, particularly the vestibular variant (previously denominated acoustic neurinomas), than neurofibromas. For some time, it was thought that patients with albinism who had CaLS and flexural freckles also simultaneously had NF1, but this possibility has been ruled out in at least 2 families in whom mutations in the KIT gene have been confirmed but with no NF1 mutations.36,37 Some authors have proposed that KIT mutations lead to altered activity of the SPRED1 gene and the development of phenotypic manifestations similar to Legius syndrome in patients with albinism.38 Ring chromosome syndromes are characterized by congenital abnormalities with facial dysmorphia, microcephalia, clinodactylies, low stature, and cognitive disorders, and the presence of multiple CaLS with typical characteristics have been reported in several of them. The recently described constitutional mismatch repair syndrome may also be associated with hypo or hyperpigmented lesions, but in the latter case, they appear to have more irregular and diffuse borders than typical CaLS.39 Despite the rarity of this syndrome, its possibility should not be ignored, as patients are at high risk of tumors in the intestines and central nervous system, as well as hematological cancers in the early years of life, and up to 97% of the 34 patients in follow-up in an international consortium study had at least one CaLS since childhood.39 Finally, other genodermatoses indicated in the table may be associated with CaLS,12 but there are usually too few of them to meet the diagnostic criterion for NF1.
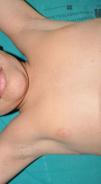
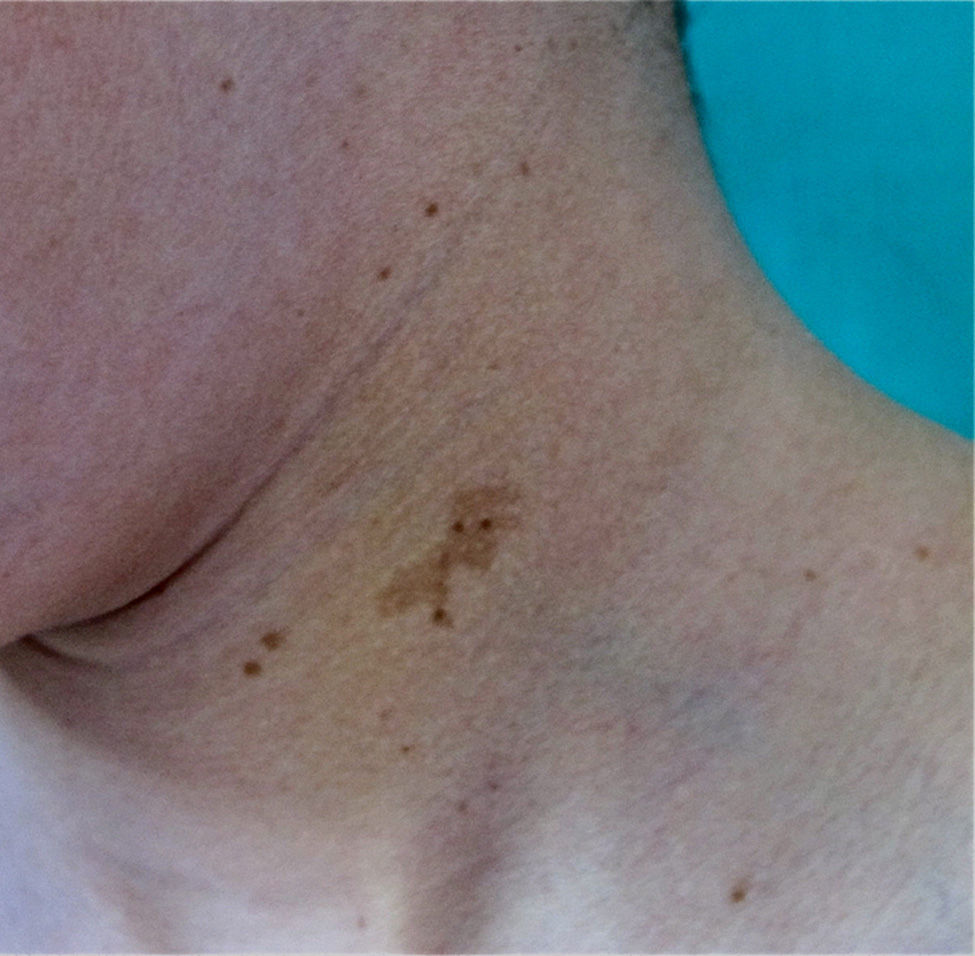
FrecklesFreckles, traditionally known as the Crowe sign, are small pigmented lesions (1 or 2mm) of light brown color that are not usually present at birth but rather develop during childhood, usually from age 2 years onwards.11,40 In exceptional cases, they may be observed congenitally, and rarely in the first few months of life.11 The presence of axillary and inguinal freckles is very characteristic, and detection satisfies 1 of the 7 diagnostic clinical criteria, but they can also be observed on other sites such as the face, neck, and trunk (Fig. 5). Their incidence is variable, ranging from 21% to 93.7%, even in series that only included pediatric patients.9,10 The number of freckles (which ranges from 2 to dozens of lesions), the size (not always millimetric), and site (sometimes away from the folds) is extremely variable, and this may explain the highly divergent prevalence estimates. The histological and ultrastructural findings are identical to those of the CaLS, and so some authors propose to include freckles and CaLS in the same diagnostic criterion.41 We also support this proposal, particularly if we remember that Legius syndrome meets 2 criteria in 48% of cases and presence of both criteria does not increase the specificity of diagnosis.24
Freckles can appear in other genetic diseases included in the differential diagnosis (Table 2), particularly in cases in which they coexist with CaLS.
NeurofibromasNeurofibromas are benign tumors that are derived from the neural sheath of the peripheral nerves. They are composed of a heterogenous mixture of Schwann cells, fibroblasts, and perineural cells, mastocytes, axons, endothelial cells, and abundant extracellular matrix. It appears that the cells that proliferate initially are Schwann cells, and it is these cells that have NF1 mutations in both alleles.42 The presence of at least 2 cutaneous neurofibromas or at least 1 plexiform neurofibroma is a diagnostic criterion for NF1. However, these lesions do not usually develop (or do not become apparent) in early childhood, and so years may pass before a patient meets this criterion.15 It is calculated that approximately 38% of patients have 1 or several types of neurofibroma before the age of 17 years.9–11
The classification of neurofibromas varies according to different authors, and this is often confusing because clinical and histological terms are mixed; in some cases they are classified as superficial, deep, and plexiform,7 whereas in others they are divided into cutaneous, subcutaneous, and plexiform (nodular and diffuse).43 In actual fact, it is difficult to classify neurofibromas with exclusively clinical or histological criteria, as some lesions are not readily classified because of a mixed infiltrative pattern, more than 1 histological component, or histological and clinical transformations during their lifetime. In addition, in any attempt at a clinical classification, the terms of the current NIH clinical criteria, used by the entire scientific community, should be kept.
Bearing the above in mind, we propose a clinical classification based on 2 large groups; superficial neurofibromas and deep neurofibromas (Table 3). Among the superficial neurofibromas, we differentiate between cutaneous neurofibromas (also called dermal neurofibromas in the literature) and subcutaneous neurofibromas, which, although they can have a dermal component, are essentially located in the subcutaneous cell tissue and may even infiltrate the deep soft tissue. Subcutaneous neurofibromas can be nodular or diffuse on palpation, whereas the deep lesions are not usually palpable and require imaging tests to be visualized. In practice, most subcutaneous neurofibromas and deep neurofibromas usually correspond to plexiform neurofibromas of the NIH clinical criteria.
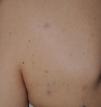
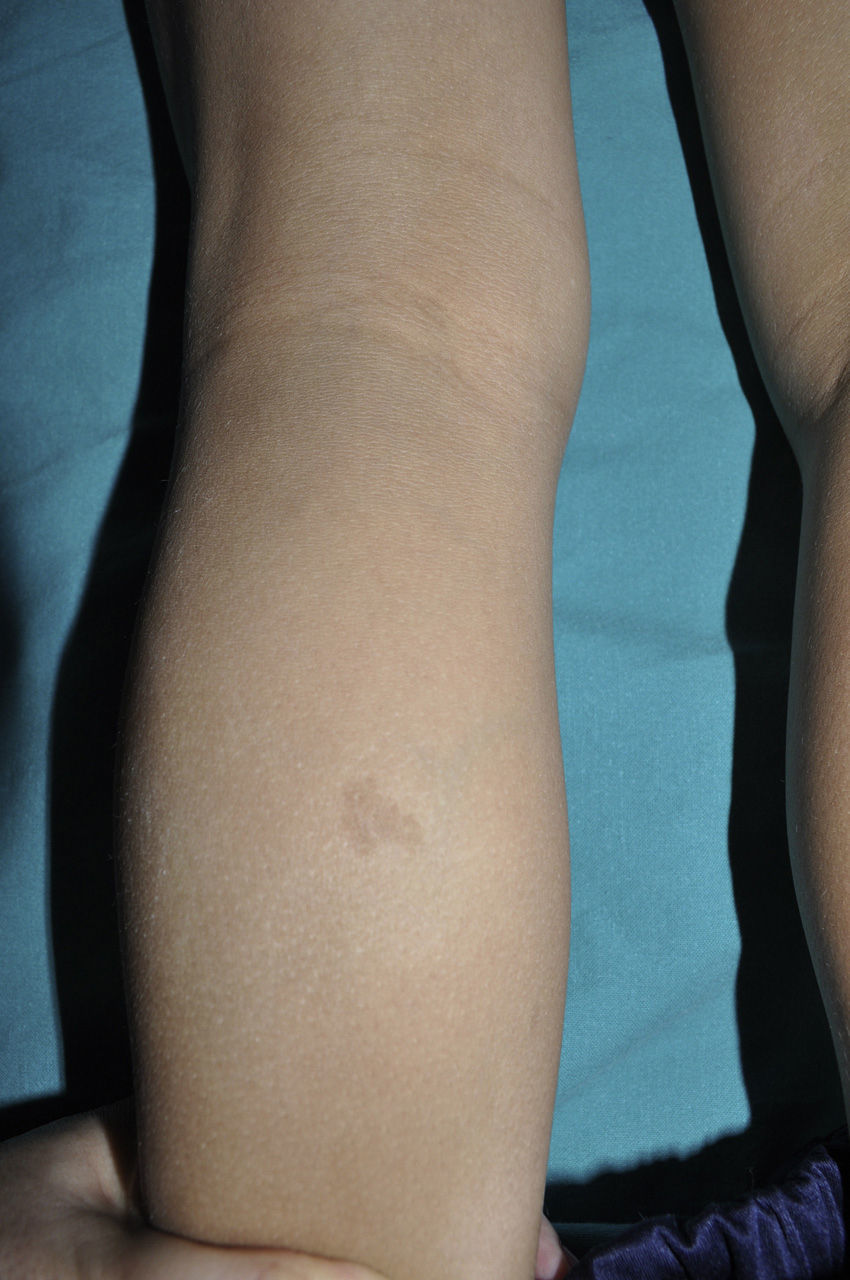
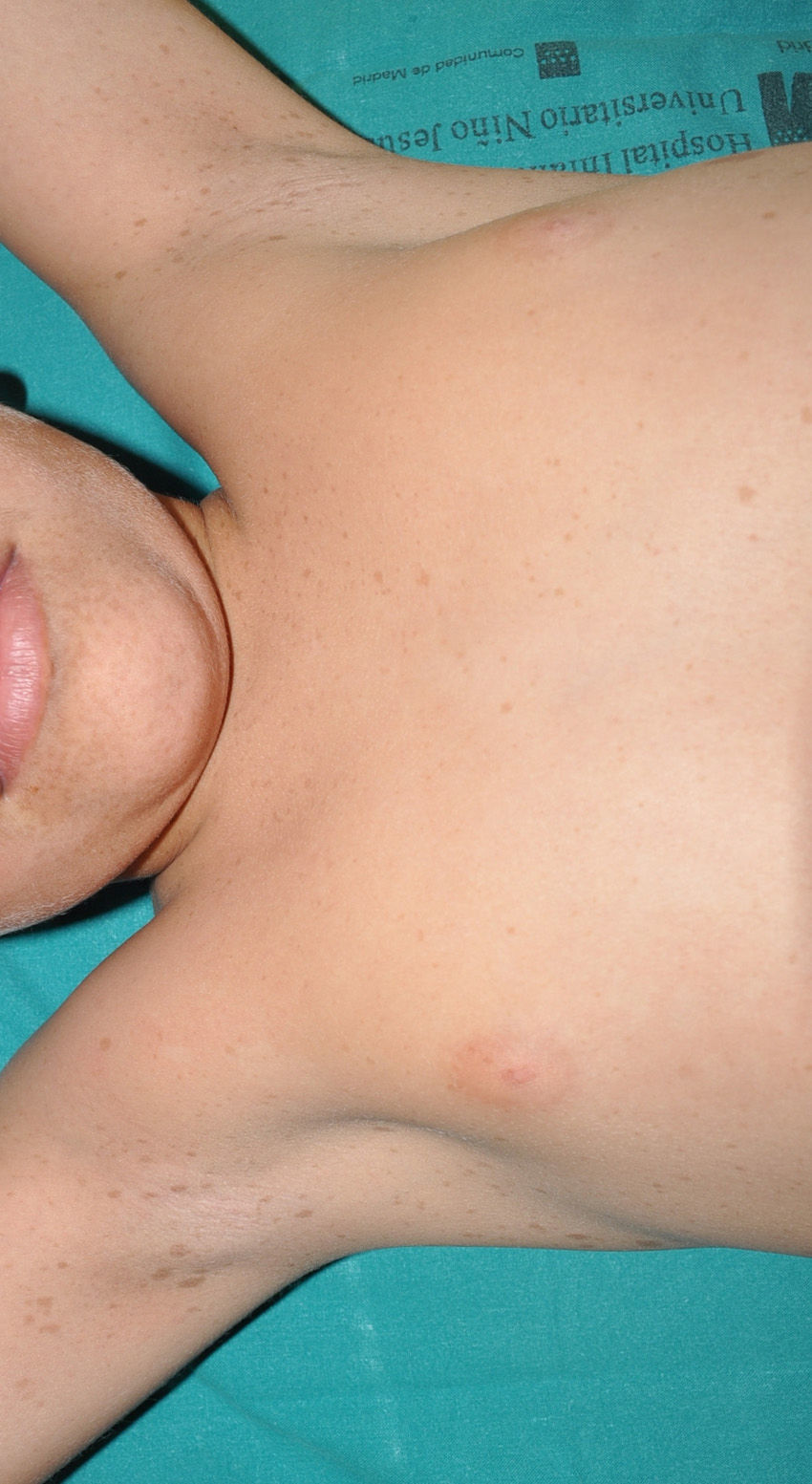
Cutaneous neurofibromas are more frequent, and usually appear from 8 years onwards, increasing in number with increasing age.13,15 In the initial phase, the cutaneous neurofibromas are almost imperceptible, hardly raised, and discretely palpable (Fig. 6). There are 2 critical periods of development, adolescence and pregnancy,44 and so these lesions are thought to be hormone dependent and, in fact, it has been shown that they have progesterone receptors.45 Their most typical morphology is one of a raised, sessile lesion of soft or elastic consistency, that is depressible on palpation (buttonholing) (Fig. 7). They can be found at any site, but they have a particular predilection for the trunk and, in the case of adult women, the periareolar region. Sometimes, they are seen as solitary lesions in healthy individuals, and at least 2 lesions must be present to be considered as a criterion for NF1. Although they are usually asymptomatic, they can induce itching, which is attributed to abundance of mastocytes that infiltrate the lesions. At times, they may need to be surgically removed when they cause esthetic or functional problems because of their number or size, although malignant transformation is very rare.43 From the histological point of view, cutaneous neurofibromas are unencapsulated tumors characterized by a mixture of Schwann cells, fibroblasts, mastocytes, and perineural cells immersed in a collagen stroma of variable myxoid component.
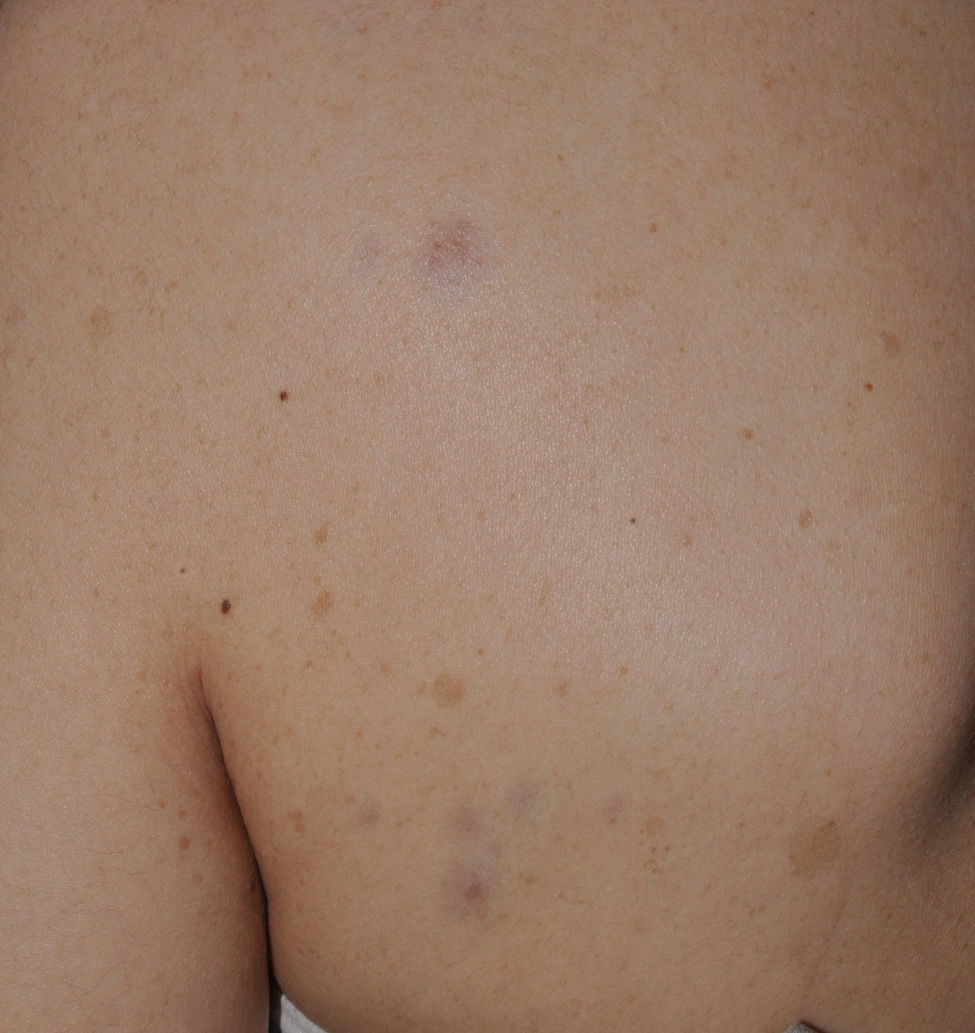
Blue-red macules and pseudoatrophic neurofibromas are 2 variants of cutaneous neurofibromas with a predilection for the trunk.46,47 Blue-red macules are lesions measuring 1-2cm in diameter with a tenuous blue-red color that are not raised (or are slightly bulging) and depressible on palpation (Fig. 8). Pseudoatrophic neurofibromas in contrast measure about 5-10cm and appear similar to normal skin, but they are slightly depressed and soft on palpation. They appear to be present in approximately 9% of adult patients.47 Although studies have not been performed in children, they probably follow the same time course as typical cutaneous neurofibromas, and we see them above all in adolescents. The subtlety of their color and/or absence of a raised border means they are often go unnoticed, but they are an important diagnostic finding for confirming the disease.48 Histologically, the blue-red macules are characterized by proliferation of neural tissue that surrounds and infiltrates the vessels of the papillary and reticular dermis, whereas the pseudoatrophic neurofibromas show decreased collagen in the reticular dermis and increased perivascular neural tissue.47,49
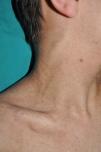
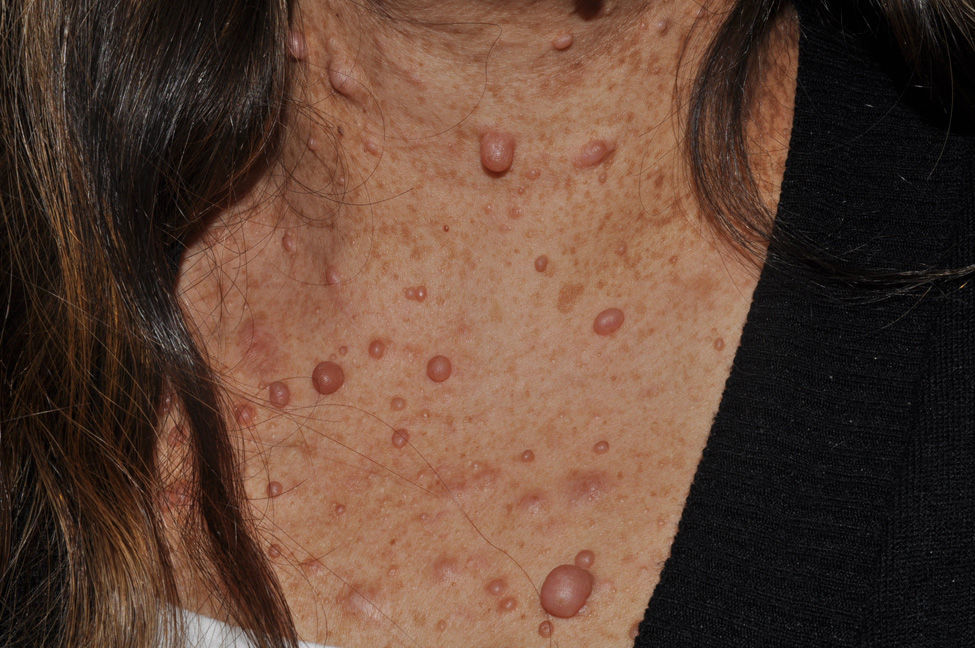
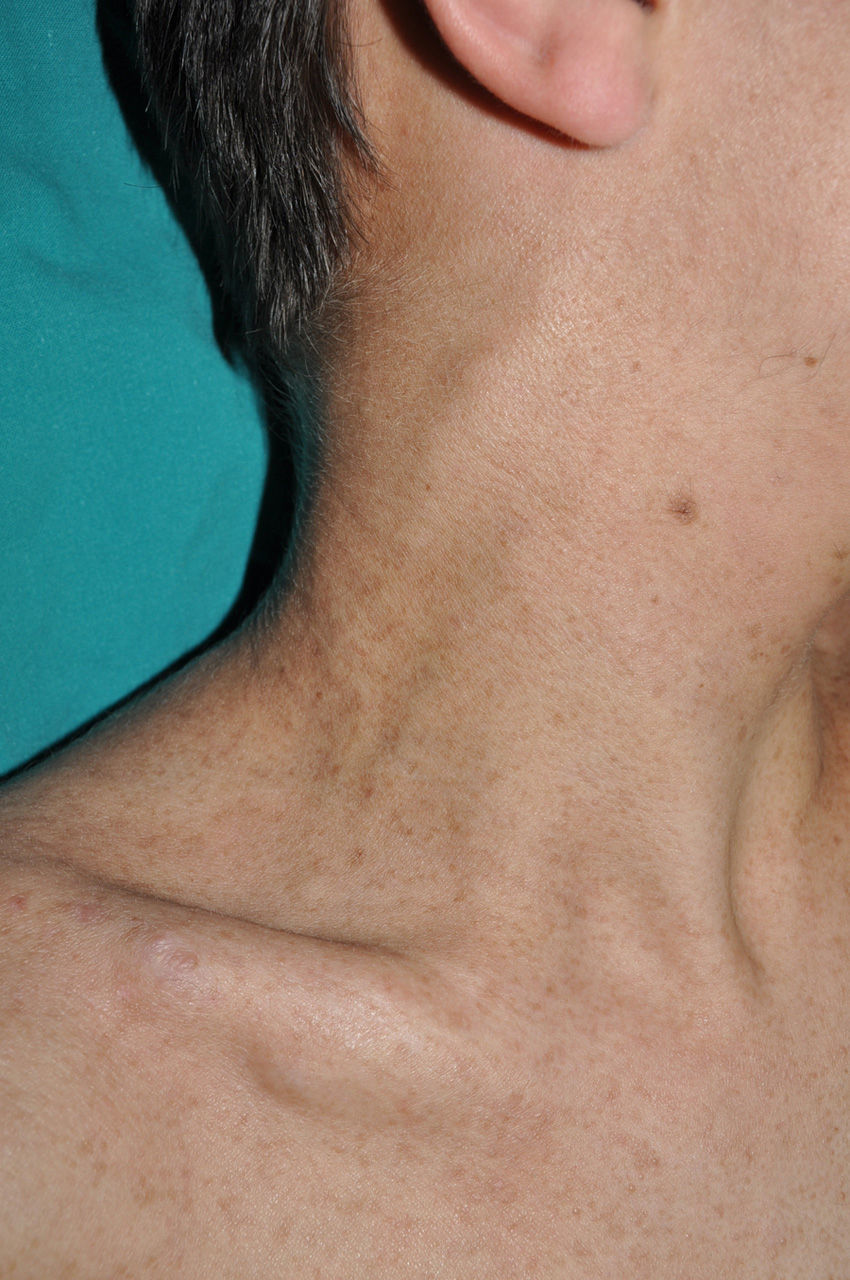
Subcutaneous neurofibromas, which we generally class as plexiform neurofibromas because they are usually associated with this histological pattern,43 can be predominantly diffuse or nodular on palpation. The diffuse lesions are manifest as subcutaneous masses of greater or lesser extension, with poorly defined borders and elastic consistency. Nodular subcutaneous neurofibromas consist of nodular lesions of rubbery consistency that follow the peripheral nerve tracts. On palpation, linear and beaded lesions with a firm consistency are apparent.7 They can appear anywhere on the body, but they are particularly common on the cephalic pole and neck, where they can be confused with swollen lymph nodes in their early phases (Fig. 9).
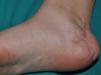
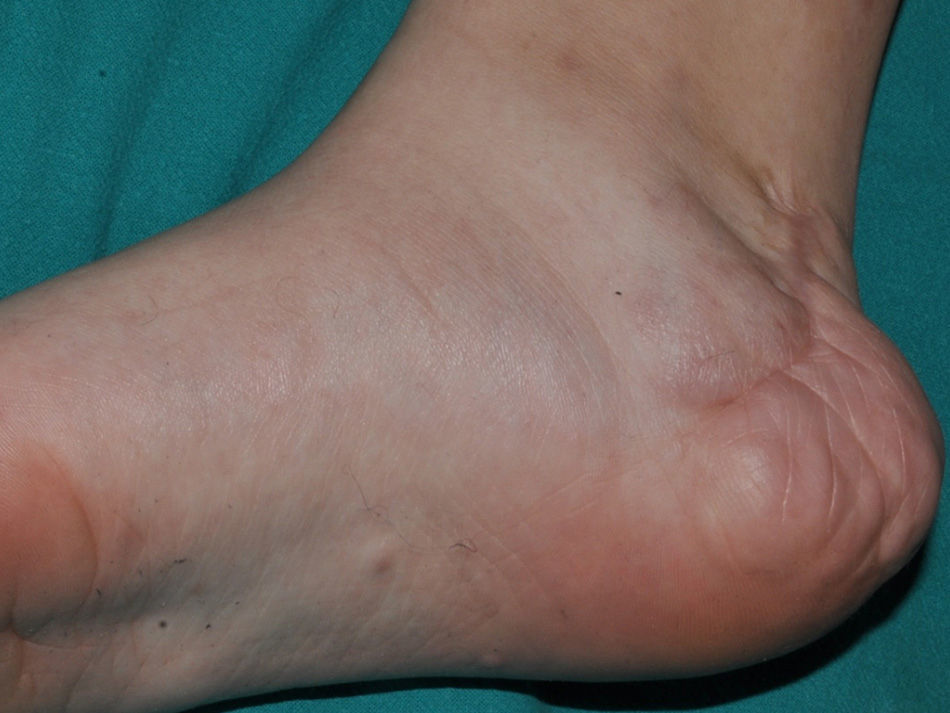
Some superficial neurofibromas may be evident at birth, and in approximately 5% of patients, underlying congenital hyperpigmentation and/or hypertrichosis can be observed (Fig. 3).9 As the children get older, the lesions become palpable and take on the typical bag-of-worms consistency of neurofibromas (Fig. 10). Early recognition of this type of neurofibroma in children less than 2 years old with CaLS is important because it is an additional clinical criterion necessary to confirm diagnosis of NF1. It is necessary to distinguish these lesions from extensive CaLS, smooth muscle hamartomas, Becker nevus, and congenital melanocytes.
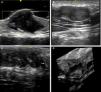
Skin ultrasound with high resolution probes (20-22MHz) enables diagnostic confirmation of superficial neurofibromas quickly and noninvasively. The presence of well-defined hypoechoic masses in the dermis (cutaneous neurofibromas in the dermis), irregular dermohypodermal regions (diffuse subcutaneous neurofibromas), or regions with oval or beaded morphology (subcutaneous nodular neurofibromas) is characteristic in subcutaneous cell tissue, with the intralesional Doppler images of showing tenuous or absent vascularization (Fig. 11).50,51
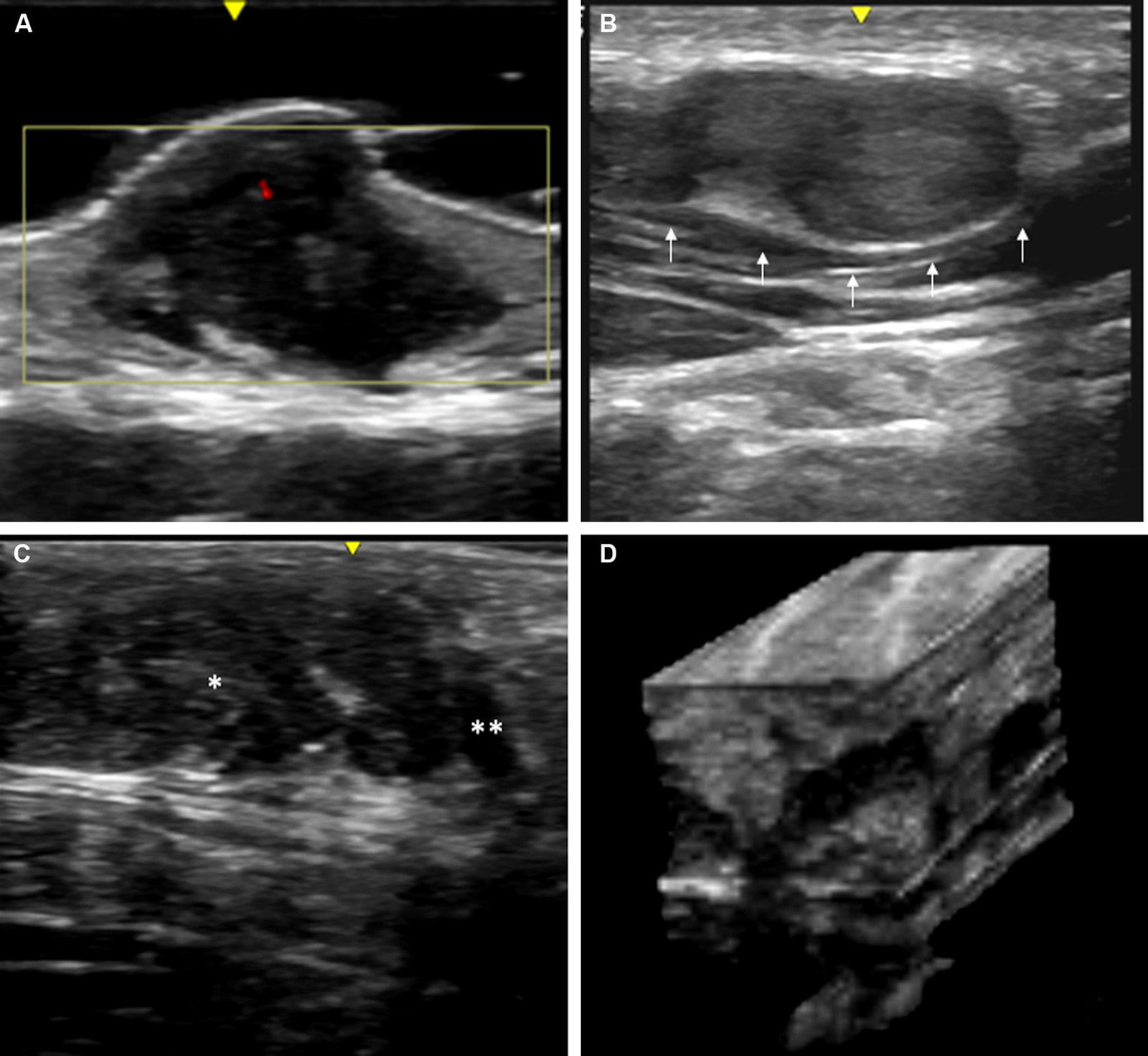
High-resolution ultrasound image (20MHz) of different neurofibromas. A. Cutaneous superficial neurofibroma where a well-delimited hypoechoic area can be seen in the dermis with a minimal Doppler signal (red color). B. Nodular subcutaneous neurofibroma of the patient in Fig. 9, in which a bilobulated, well-delimited, heteroechogenic lesion can be seen with mild posterior reinforcement that follows the path of a laterocervical peripheral nerve. C. Subcutaneous neurofibroma that occupies the subcutaneous cell tissue and deep dermis in which diffuse areas (*) and chordal areas (**) can be observed. D. Three-dimensional ultrasound reconstruction in which a convoluted linear area can be seen associated with thickening of a peripheral nerve.
Deep (or internal) neurofibromas are not usually directly related to the skin, often go unnoticed in clinical examination, are usually present from birth, have delayed development, and become symptomatic, and so are not detected until later. They may have a nodular or more diffuse morphology and present at any site. This will determine whether they are symptomatic and what associated morbidity is present. The size increases from adolescence onwards and, although this change is more marked in females, no hormonal markers have been found to explain the change.44,52 Several studies, including our series of patients, have shown a prevalence of these lesions in children of around 10%.9,13 Given that 8% to 13% of these neurofibromas may undergo malignant transmformation,43 and they cannot be detected in clinical examination, indicators of their presence have been sought. It has been observed that young adults (aged 30 years or younger), with hardly any cutaneous manifestations of NF1 (that is, fewer than 6 CaLS and no cutaneous neurofibromas) but with 2 or more subcutaneous neurofibromas are at greater risk of internal neurofibromas and this should be taken into account during follow-up.53 Although some authors have detected plexiform neurofibromas in 57% of patients who underwent full-body magnetic resonance imaging,54 there are currently no guidelines as to which patients should be monitored with these imaging studies.
There are 2 varieties of NF1 that are principally characterized by the presence of deep neurofibromas: spinal neurofibromatosis, in which deep bilateral neurofibromas are present in all spinal roots with or without other manifestations of NF1,55 and orbital or cranio-orbitotemporal neurofibromatosis, in which the neurofibroma occupies the entire orbit, invades the orbital muscles, and is associated with exophthalmos, ocular assymetry, temporal deformity, sphenoid dysplasia, and temporal lobe herniation.56 This latter form of NF1 is very disfiguring and very difficult to correct.
From the histological point of view, neurofibromas can be myxoid (many of the dermal variants), diffuse (with stroma that is more collagenized than myxoid and infiltrating) or plexiform, a variant in which a tortuous thickening of the peripheral nerves is observed and which is considered pathognomonic of NF1. However, relatively frequently, the diffuse histological and plexiform pattern coexist in subcutaneous and deep lesions,57 and so the clinical nomenclature does not always coincide with the histological characteristics.
Massive, growing neurofibromas, for which surgery is impractical, are responsible for significant morbidity in patients with NF1. Clinical trials are ongoing,58 and animal models are under development to assess the efficacy of MEF1/2 inhibitors in these tumors.54
In contrast to typical pigmented lesions of NF1, which in themselves are not pathognomonic, no other disease with CaLS and freckles is also associated with neurofibromas. Massive subcutaneous neurofibromas may require differential diagnosis with other syndromes (Table 2).
In conclusion, CaLS and freckles are the most widely recognized manifestations of NF1, but they are not pathognomonic and are insufficient for definitive diagnosis in the early years of life. Given that neurofibromas do not develop (or do not become apparent) until children are of school age, it is reasonable to recommend conducting regular follow-up of these patients, ruling out the presence of other noncutaneous diagnostic criteria, and considering other possible diagnoses.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
We would like to thank Francisco Javier García Martínez, dermatologist of the Hospital del Sureste, Arganda, Madrid, for his help in taking and describing the ultrasound images of the superficial neurofibromas.
Please cite this article as: Hernández-Martín A, Duat-Rodríguez A. Neurofibromatosis tipo 1: más que manchas café con leche, efélides y neurofibromas. Parte I. Actualización sobre los criterios dermatológicos diagnósticos de la enfermedad. Actas Dermosifiliogr. 2016;107:454–464.


