



INTRODUCCIÓN
La miofibromatosis infantil fue descrita por Stout en 1954 como «fibromatosis generalizada congénita»1. En 1981, Chung y Enzinger revisaron 61 casos del Armed Forces Institute of Pathology (AFIP) y propusieron la de-nominación de miofibroma y miofibromatosis respectivamente para los casos solitarios y múltiples2, basándose en la apariencia y los rasgos inmunohistoquímicos de las células tumorales. Aunque es una entidad poco frecuente, probablemente no es tan rara como se pensaba: hasta 1997 se encuentran al menos 231 casos publicados3, 4. Se trata de un trastorno mesenquimal en el que aparecen tumores que afectan a piel, tejido celular subcutáneo, músculos, huesos y/o vísceras. El número de tumores puede variar desde 1 a más de 100. El 80% de los casos son congénitos y es raro que comience después de los 2 años de edad5. No obstante, hay una forma del adulto que suele ser solitaria6. La tendencia natural de los tumores es hacia la involución espontánea7, pero la presencia de afectación visceral ensombrece el pronóstico8.
DESCRIPCIÓN DEL CASO
Se trata de una niña de 23 días de edad que fue remitida a nuestro servicio por la presencia desde el nacimiento de varias lesiones cutáneas de aspecto tumoral que afectan tronco y extremidades. La niña era hija de padres sanos y fruto de un embarazo y parto normales. Había presentado al nacimiento un bajo peso para la edad gestacional (2.070 g), una talla de 43 cm y un perímetro cefálico de 32 cm. A la exploración se observaban dos lesiones papulosas, de color rojizo y consistencia fibrosa en el brazo y muslo izquierdos (fig. 1). Además presentaba dos nódulos subcutáneos en el abdomen, próximos a los flancos, recubiertos de piel de aspecto normal. El resto de la exploración física no reveló alteraciones, salvo la presencia de una parálisis facial derecha.
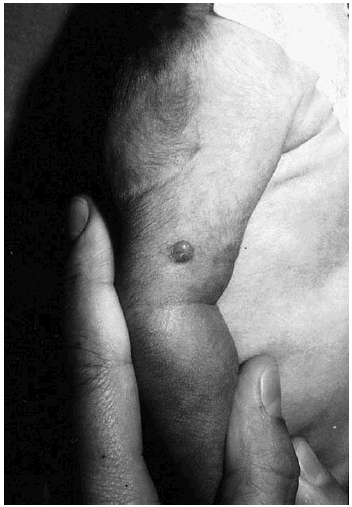
Fig. 1.--Pápula de consistencia fibrosa en el brazo.
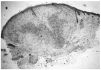
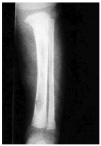
Se realizó biopsia de uno de los tumores que mostró una lesión nodular bien definida, no encapsulada, localizada en la dermis y respetando la epidermis (fig. 2). La parte periférica de la lesión se componía de fascículos compuestos por células fusiformes, de amplio citoplasma eosinófilo y con núcleos alargados. En el centro de la tumoración se encontraban cúmulos de cé-lulas redondeadas o poligonales, con citoplasmas de bordes mal definidos y núcleos hipercromáticos. Esta zona mostraba espacios vasculares con luces interconectadas en un patrón en «asta de ciervo» (fig. 3). La inmunohistoquimia demostró positividad para vimentina y actina específica del músculo. Se practicó hemograma y bioquímica sérica, que fueron normales. La radiografía de tórax, la ecografía abdominal y cardíaca y la tomografía axial computarizada (TAC) cerebral no detectaron anomalías. En la serie ósea se detectaron múltiples imágenes líticas diafisometafisarias en fémures, tibias y húmeros, así como en mandíbula y pelvis, con bordes bien definidos no escleróticos (fig. 4). Se realizó una interconsulta a Neurología solicitando valoración de la parálisis facial, y ésta fue etiquetada de parálisis facial periférica de etiología probablemente obstétrica.
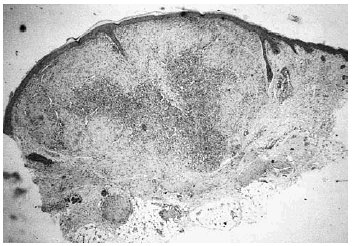
Fig. 2.--Lesión tumoral intradérmica que muestra un patrón bifásico.
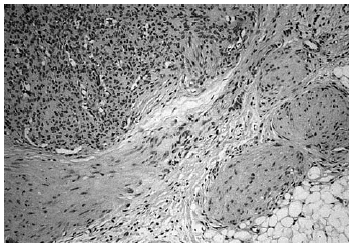
Fig. 3.--Detalle histológico. En la parte superior izquierda: patrón hemangiopericitoide. En la parte inferior derecha: patrón leio-miomatoide.
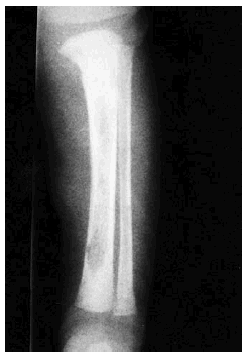
Fig. 4.--Imagen lítica en la tibia.
A los 5 meses de edad la niña se encuentra bien, con adecuado desarrollo ponderoestatural. Las lesiones cutáneas han mejorado discretamente, pero persiste la parálisis facial.
DISCUSIÓN
La miofibromatosis infantil puede clasificarse en solitaria o múltiple, atendiendo al número de lesiones presentes. Tanto las formas solitarias como las múltiples tienen preferencia por el sexo masculino en una proporción de 2 a 19, 10. Las formas solitarias son las más frecuentes, suponen del 50% al 70% de los casos y afectan generalmente a la piel o al tejido celular subcutáneo, con menor frecuencia al músculo o al hueso y raramente a vísceras9. Las formas múltiples pueden tener más de 100 tumores en total y se clasifican a su vez en dos grupos. En el primero se afecta piel, tejido celular subcutáneo, músculos y/o huesos, pero no vísce-ras, y el pronóstico es favorable. En el otro, que supone el 25% de las formas múltiples, hay afectación visceral, lo que implica hasta un 75% de mortalidad4. En este grupo los órganos más frecuentemente afectados son el corazón, el pulmón, el tracto gastrointestinal, el hígado y el páncreas. La muerte habitualmente se debe a complicaciones cardiopulmonares o gastrointestinales11. Parece que el órgano cuya afectación implica peor pronóstico es el pulmón.
En la piel los miofibromas suelen aparecer predominantemente en cabeza y tronco, y menos veces en extremidades. Se presentan como nódulos asintomáticos de consistencia gomosa de entre 0,5 y 7 cm de diámetro, recubiertos de piel normal o rojiza. Se han descrito formas en placa12 y formas atróficas; estas últimas se suponen debidas a la involución de casos de desarrollo precoz intrauterino8, 13. Aunque los tumores tienden a involucionar espontáneamente, hay casos en que persisten y a veces aparecen nuevas lesiones posteriormente9, 14, por lo que es importante hacer seguimiento de estos pacientes.
Histológicamente la miofibromatosis infantil presenta un patrón bifásico característico2, 15. Suelen aparecer uno o varios nódulos bien definidos, no encapsulados, en dermis o tejido celular subcutáneo. La periferia de cada nódulo se compone de fascículos de células fusiformes, de citoplasma eosinófilo, con núcleos alargados, a veces en cigarro puro, que recuerdan al leiomioma. En cambio, el centro se compone de células menos diferenciadas, poligonales, de núcleos hipercromáticos redondeados u ovales que delimitan espacios vasculares interconectados en un patrón muy similar al del hemangiopericitoma. Ocasionalmente, el patrón histológico se invierte, de forma que la zona similar al hemangiopericitoma se dispone en la periferia y la zona similar al leiomioma es central16. Inmunohistoquímicamente se detecta positividad para actina y vimentina y negatividad para desmina y proteína S-100. Por las características ultraestructurales de las células y el patrón inmunohistoquímico se acepta que es un tumor de estirpe miofibroblástica2. No obstante, otros autores encuentran evidencia de diferenciación hacia músculo liso14 o hacia pericito17. La similitud histológica con el hemangiopericitoma congénito y con el fibrosarcoma congénito puede plantear problemas de diagnóstico diferencial, ya que también tienen semejanzas clínicas y afectan al mismo grupo de edad17. El fibrosarcoma tiene peor pronóstico y es importante distinguirlos, para lo cual puede ser útil realizar estudios citogenéticos. El fibrosarcoma presenta anomalías cromosómicas numéricas, generalmente trisomías, que no aparecen habitualmente en la miofibromatosis18. Con respecto al hemangiopericitoma congénito, se ha propuesto que se trate de una forma de miofibromatosis infantil en la que predomina el patrón histológico similar al hemangiopericitoma19.
La etiología de la miofibromatosis es controvertida. La aparición de la mayoría de los casos en niños recién nacidos plantea la posibilidad de algún factor causal de acción intrauterina4, 11. En ensayos experimentales con cobayas tratados con estrógenos se ha conseguido desarrollar tumores similares a los de la miofibromatosis20. Sin embargo, estas hipótesis no explican los casos de aparición tardía. La presencia de casos familiares apoya una etiología genética con un patrón de herencia dominante con penetrancia reducida21, 22.
La mayoría de los casos de miofibromatosis no requiere tratamiento. Se puede hacer cirugía de los casos solitarios, siendo bajo el porcentaje de recidivas 8.En casos seleccionados de gran afectación funcional, irresecables o con afectación visceral se puede complementar el tratamiento con radioterapia y/o quimioterapia. Se han utilizado combinaciones de vincristina, adriamicina-D y ciclofosfamida, así como interferón-α, con resultados dispares7, 8.
La evaluación de un niño con miofibromatosis debe incluir, además de una biopsia confirmatoria, una exploración física completa e historia familiar. Asimismo es obligada la realización de una serie ósea. La ecografía abdominal y la placa de tórax se pueden sustituir por una TAC o mejor aún por una resonancia magnética (RM), ya que esta última tiene mayor resolución para este tipo de tumores23. Además debe realizarse un seguimiento de los pacientes por la posibilidad de desarrollar nuevos tumores y dar consejo genético a los padres.


