







INTRODUCCION
El miofibroma cutáneo del adulto es una entidad de reciente descripción1 que puede considerarse como la variante solitaria y benigna de la miofibromatosis infantil2. A tenor de las series publicadas en los últimos años1, 3-6, probablemente se trate de una lesión relativamente frecuente.
En el adulto el miofibroma cutáneo se manifiesta como un nódulo único de consistencia firme, sin tendencia a la involución6, a diferencia de lo que ocurre en los casos infantiles. Histopatológicamente se caracteriza por presentar áreas hialinizadas, de aspecto «miofibroblástico», entremezcladas con otras de aspecto hemangiopericitoide6. Aunque existe cierta controversia al respecto, los últimos artículos publicados proponen un origen a partir de componentes miofibroblásticos o miopericitarios de las paredes vasculares3, 6, 7.
Presentamos un caso de miofibroma cutáneo del adulto en su variante multinodular y realizamos una revisión de la literatura al respecto.
DESCRIPCION DEL CASO
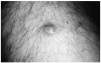
Se trata de un hombre de 73 años que consultó por una lesión en cara anterior del muslo derecho de 10 años de evolución. Durante este tiempo la lesión había permanecido estable y era ligeramente dolorosa a la manipulación. En la exploración se observaba un nódulo multilobulado, de consistencia pétrea, de 2 cm de diámetro. La piel suprayacente era aparentemente normal aunque mostraba una coloración eritematoamarillenta (fig. 1). El resto de la exploración física no demostró ninguna otra anomalía, salvo una adenopatía submandibular.
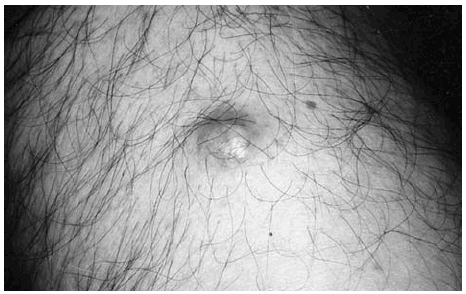
Fig. 1.--Aspecto clínico de la lesión: nódulo eritematoso amarillento multilobulado de 2 cm de diámetro, pétreo a la palpación. Localizado en la cara anterior del muslo izquierdo.
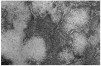
Se realizó una extirpación quirúrgica de la lesión. El estudio histopatológico mostró una tumoración dérmica multilobulillar con una zona central hipocelular, esclerosa y otra hipercelular en la periferia en la que se veían multiples espacios vasculares (fig. 2). Estas áreas más periféricas de espacios vasculares ramificados recordaban a un hemangipericitoma, y se continuaban insensiblemente con nódulos esclerosos. También se observaban nódulos hemangiopericitoides a distancia y sin conexión aparente con la tumoración principal (fig. 3).
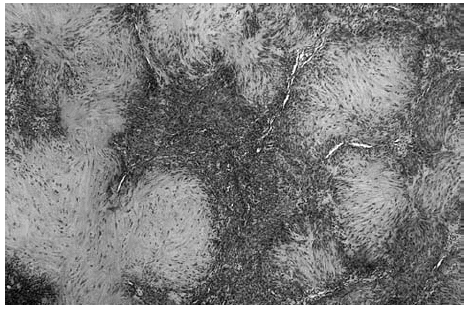
Fig. 2.--Panorámica del tumor situado en dermis, con áreas esclerosas hipocelulares junto a otras hipercelulares. Destaca la presencia de hendiduras vasculares en la periferia de algunos nódulos.
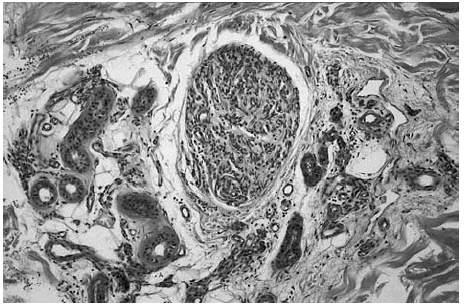
Fig. 3.--Detalle de un nódulo de aspecto hemangiopericitoide que contiene múltiples espacios vasculares. Localizado a cierta distancia de la tumoración principal.
Las áreas hipocelulares presentaban un estroma escleroso. Las células que contenían se disponían en haces no muy bien estructurados. Estas células mostraban un núcleo alargado con extremos redondeados que recordaban fibras musculares lisas (fig. 4).
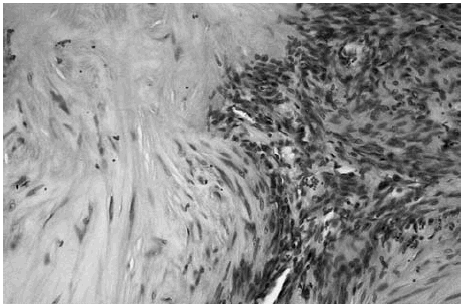
Fig. 4.--Detalle de una transición entre una zona hipocelular con células fusiformes dispuestas en haces no muy bien estructurados con un estroma hialinizado y una zona hipercelular.
Las zonas más celulares presentaban múltiples espacios vasculares, algunos de los cuales contenían un único eritrocito. Rodeando estos espacios vasculares se observaban células de núcleo redondeado, con límites citoplasmáticos poco evidentes. En algunas zonas, estas células se disponían de forma angiocéntrica (fig. 5). En la periferia del tumor o a distancia del mismo se observaban áreas con evidente patrón hemangiopericitoide constituido por espacios vasculares que se ramificaban de forma que recordaban astas de ciervo (fig. 6).
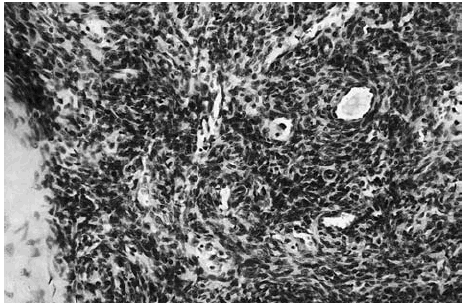
Fig. 5.--Detalle de un nódulo hipercelular en el que además de múltiples espacios vasculares que contenían escasos hematíes se apreciaba un vaso de mayor calibre alrededor del cual se disponen las células.
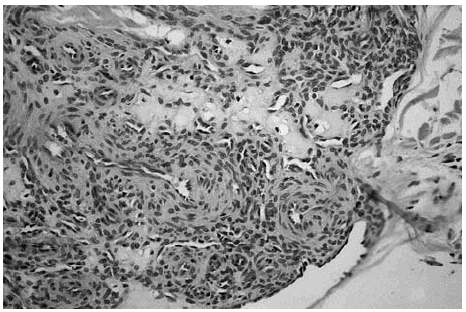
Fig. 6.--Detalle de un área hemangiopericitoide que muestra espacios vasculares que en zonas, recuerdan astas de ciervo. Se aprecia también una hendidura vascular de mayor tamaño en la periferia del nódulo.
Muchos de los nódulos, tanto hipo como hipercelulares, presentaban una hendidura periférica en media luna.
Tras 2 años de seguimiento no ha habido recidiva.
DISCUSION
A pesar de que la miofibromatosis infantil había sido descrita previamente por Stout en 1954, fueron Chung y Enzinger2 en 1981 quienes la independizaron como entidad al describir una serie de 61 casos. Estos autores citaron brevemente la existencia de casos en adultos caracterizados por lesiones solitarias, sin tendencia a la regresión y sin afectación visceral.
En 1989, Daimaru et al1 describieron específicamente el primer caso de miofibroma cutáneo en un adulto, pero fueron Beham et al3 en 1993 quienes separaron este tipo de lesiones en dos entidades, la infantil y la del adulto, basándose principalmente en su comportamiento biológico distinto. Kutzner et al4 y Requena et al6 publicaron en 1996 la mayor serie de casos de miofibroma cutáneo del adulto y postularon un origen pericitario de la tumoración. Similares conclusiones fueron posteriormente publicadas por Granter et al7. Mentzel et al8 habían propuesto la misma histogénesis en los casos de miofibromatosis infantil 2 años antes.
Clínicamente1 se trata de un nódulo único, a excepción de uno de los casos de Requena et al, que tenía siete lesiones. Parece que el miofibroma cutáneo del adulto es algo más frecuente en mujeres que en hombres3, aunque se precisan series más amplias para confirmar este dato. Se ha descrito en pacientes de hasta 77 años9. El límite de edad inferior es de 12 años, ya que los casos de edades inferiores se consideran infantiles10 de una forma arbitraria y probablemente errónea. La lesión suele ser multilobulillar, aunque circunscrita, de consistencia dura, pétrea o gomosa9, 10. La piel suprayacente puede ser de aspecto normal , de apariencia vascular, o adoptar una coloración amarillenta11. Un caso mostró una lesión ulcerada3. El tamaño oscila entre 0,5 y 3 cm de diámetro3 y en el momento del diagnóstico suele haber tenido una evolución larga1. En la mayoría de casos es asintomático, aunque no faltan las referencias de pacientes en los que era doloroso a la palpación6. Se localiza, por orden decreciente de frecuencia, en extremidades, cabeza y cuello y tronco4, 6, aunque también se han descrito en lugares tan dispares como pene11, conducto auditivo externo12 y en varias ocasiones en cavidad oral1, 3, 9.
Los hallazgos histopatológicos1, 3 son característicos. Se trata de un tumor localizado en dermis reticular con un patrón bifásico: áreas de apariencia miofibroblástica constituidas por células fusiformes con núcleos alargados de extremos romos que recuerdan fibras musculares lisas, dispuestas habitualmente en haces inmersos en un estroma escleroso; y áreas de apariencia hemangiopericitoide, constituidas por espacios vasculares ramificados rodeados por células de apariencia pericitoide o glomoide, a veces con disposición angiocéntrica. A veces se observa una hendidura en forma de media luna en la periferia de los nódulos.
Lás áreas hemangiopericitoides se localizan habitulamente en el centro en los casos infantiles, mientras que en los casos de los adultos su disposición es mucho más variable. En ocasiones se ha observado una aparente invasión vascular con protrusión de nódulos celulares en las luces vasculares, sin que ello tenga trascendencia desde el punto de vista pronóstico3. No se aprecian atipias ni mitosis9.
Kutzner et al4 y Requena describieron cuatro variantes histopatológicas probablemente en relación con el tiempo de evolución de la lesión: vascular, nodular, multinodular o bifásica y fascicular. De acuerdo con estos autores la variante vascular correspondería a las lesiones más jóvenes y las de más tiempo de evolución mostrarían la variante fascicular, mientras que las variantes nodular y multinodular corresponderían a las fases intermedias, aunque serían las más características y frecuentes.
El estudio inmunohistoquímico puede ayudar al diagnóstico en los casos dudosos, con un patrón característico, ya que las células neoplásicas muestran negatividad para proteína S-100 y desmina, y positividad para la actina de músculo liso, actina muscular específica y vimentina1, 6.
Desde le punto de vista ultraestructural las células neoplásicas muestran filamentos intracitoplasmáticos en las áreas miofibroblásticas además de abundante retículo endoplasmático rugoso y vesículas de micropinocitosis3, 6, 9. Estos hallazgos apoyan la naturaleza miofibroblástica5 o miopericitaria4, 6, 7 de la lesión. En la actualidad, el pericito es considerado una célula madre con capacidad para diferenciarse hacia células miofibroblásticas y células de músculo liso13, considerándose el miopericito una célula intermedia entre un pericito y una célula de músculo liso14.
El tratamiento del miofibroma cutáneo del adulto consiste en la exéresis quirúrgica. Los escasos casos descritos de recurrencias12, 13 probablemente sean debidos a una extirpación incompleta.
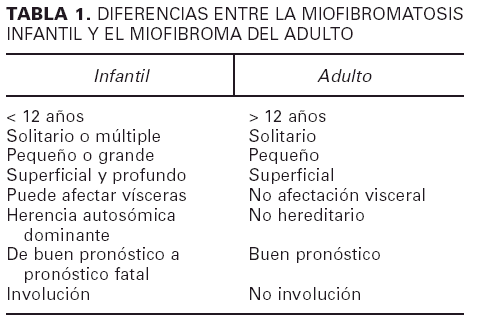
A pesar de sus similitudes, existen diferencias importantes entre la variante infantil2, 15 y la del adulto3, sobre todo a nivel de su comportamiento biológico, lo que justifica su consideración como dos entidades separadas (tabla 1). Aunque los casos infantiles muestran tendencia a la involución, cuando existen lesiones múltiples con afectación visceral el pronóstico es muy malo y la mayoría de los pacientes fallecen como consecuencia de la insuficiencia del órgano afecto. Sin embargo, la mayoría de los casos infantiles se presentan como lesiones solitarias. En ocasiones se reconoce un carácter hereditario del cuadro.
Nuestro caso corresponde a un miofibroma cutáneo del adulto en la variante multinodular. Sin embargo, presentaba la particularidad de la existencia de nódulos a cierta distancia de la lesión principal, hallazgo que no hemos encontrado descrito con anterioridad. La existencia de nódulos a distancia de la masa principal podría explicar la recidiva de este tumor en los escasos pacientes en que esto ha ocurrido.


