


INTRODUCCION
Las fibromatosis constituye un grupo de lesiones proliferativas no neoplásicas que comprende un 12% de los tumores de partes blandas de la población pediátrica1. El término fibromatosis generalizada congénita fue introducido por Stout en 19542. En 1981, Chung y Enzinger3 emplearon por primera vez la denominación miofibromatosis infantil, describiendo las características clinicopatológicas del cuadro. La revisión más amplia de casos publicados corresponde a Wiswell et al4.
Describimos un caso de miofibromatosis infantil solitaria, de aparición en el período neonatal en la que se descartó afectación multiorgánica tras estudio de extensión.
DESCRIPCIÓN DEL CASO
Se trata de una niña de 9 meses de edad sin antecedentes personales de interés. Presentaba desde los 4 meses de edad una lesión nodular, de 2 cm de diámetro máximo, situada en región deltoidea posterior izquierda. La lesión era de superficie lisa, eritematoviolácea, dura e infiltrada, pero desplazable sobre planos profundos (fig. 1). La lesión había aumentado progresivamente de tamaño hasta 2 meses antes de la consulta, en que parecía haberse estabilizado. Se extirpó quirúrgicamente.
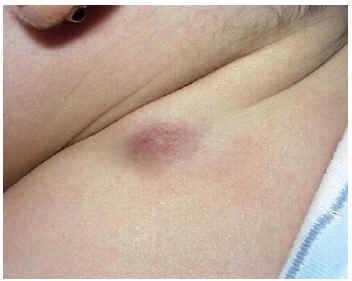
Fig. 1.--Lesión nodular infiltrada de 2 cm en región deltoidea posterior izquierda.
El estudio histopatológico mostró a nivel de dermis y tejido celular subcutáneo una lesión tumoral relativamente circunscrita, aunque de límites imprecisos, en la que se apreciaba una arquitectura zonal. En la periferia había una proliferación de células fusiformes, dispuestas en fascículos entrelazados o arremolinados, de patrón leiomiomatoso, separados por finas bandas de colágeno; en la zona intermedia, células de pequeño tamaño, separadas por espacios vasculares irregulares, de patrón hemangiopericitoide; finalmente existía una zona central de hialinización (fig. 2). No existía atipia celular, siendo infrecuentes las mitosis. En la periferia, la tumoración mostraba tendencia a infiltrar el tejido adiposo, así como los fascículos musculares presentes en el borde profundo. Asimismo se apreciaban imágenes de crecimiento intravascular.
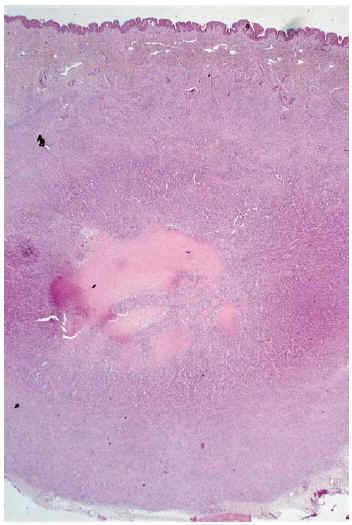
Fig. 2.--Lesión nodular en dermis y tejido celular subcutáneo con tres áreas histológicamente distintas: zona periférica de patrón leiomiomatoso, zona intermedia de patrón hemangiopericitoide y zona central hialinizada.
Se realizaron técnicas de inmunohistoquímica, apreciándose expresión en las células proliferantes de vimentina y actina, muy escasa de p53, reactividad para CD34 únicamente en los vasos, y negatividad para citoqueratinas (AE1/AE3), desmina y proteína S-100.
El diagnóstico fue de miofibromatosis infantil. Se practicó estudio de extensión para descartar afectación visceral: examen físico completo, electrocardiograma, ecografía cardíaca y abdominopélvica, radiografía de tórax y serie ósea. No se apreciaron imágenes patológicas, por lo que el diagnóstico final fue de miofibromatosis infantil solitaria. En los 12 meses de seguimiento no ha presentado recurrencia de la lesión.
DISCUSIÓN
La miofibromatosis infantil fue descrita por Stout en 19542. Caracterizada por Chung y Enzinger en 19813, constituye una proliferación benigna, probablemente hamartomatosa5, de miofibroblastos, células que muestran miofilamentos intracitoplasmáticos en el estudio ultraestructural3, 4. Fletcher et al en 1987 sugirieron que la histogénesis de la lesión correspondía al tejido muscular liso y propusieron el término de leiomioblastoma6.
El 60% de las lesiones aparecen en el recién nacido, y el 88% antes de los 2 años de edad3, y constituye el tumor fibroso más frecuente en toda la infancia4. Se han descrito casos en la edad prepuberal7, e incluso en adultos8-11. La mayoría de los casos son esporádicos, pero se han descrito casos familiares, generalmente con herencia autosómica dominante12.
Existen dos variantes de miofibromatosis infantil: a) forma solitaria (la más frecuente), que suele afectar a piel, partes blandas o hueso13, y b) forma multicéntrica, que aunque frecuentemente están limitadas a tejidos blandos y huesos, presenta afectación visceral hasta en el 37% de los casos14. Los órganos más comúnmente afectados son hueso, pulmones, sistema gastrointestinal y corazón1, 4. La forma multicéntrica con afectación visceral es denominada por muchos autores fibromatosis generalizada congénita, distinguiéndola de la fibromatosis multifocal congénita (sin afectación visceral).
Las formas solitarias tienen predilección por el sexo masculino, mientras que las formas múltiples suelen mostrar preferencia por el sexo femenino3.
Sin una clínica característica, suelen aparecer como lesiones cutáneas nodulares únicas y bien delimitadas, firmes, asintomáticas, entre 0,5 y 10 cm de tamaño15, 16, que aparecen preferentemente, y por este orden, en cabeza, cuello o tronco3. Pueden recordar a hemangiomas cuando son superficiales por su abundante vascularización16.
Histológicamente se caracterizan por la presencia de nódulos bien delimitados, con localización preferente en dermis profunda y tejido celular subcutáneo17, pudiendo estar el músculo esquelético y el hueso infiltrados por la proliferación celular16, 18. La lesión presenta un patrón bifásico, constatándose áreas fusocelulares periféricas y zonas internas con patrón hemangiopericitoide, que puede estar centrada por áreas de hialinización. No son infrecuentes tampoco los focos de necrosis, calcificación e incluso osificación4.
Los elementos que proliferan expresan vimentina y actina, en tanto que son negativos para desmina y proteína S-10015, 19.
En nuestra opinión, ante una lesión confirmada, debe realizarse estudio de extensión para descartar afectación sistémica/visceral4, 16. Debe incluir: exploración física completa, electrocardiograma, ecografía cardíaca y abdominal, radiografía torácica, serie ósea y tomografía axial computarizada (TAC) toracoabdominal (que en nuestra paciente no se realizó por su corta edad y al ser negativas el resto de pruebas). Sin embargo, otros autores7 creen que sólo habría que hacer estudio si se sospecha afectación múltiple.
Las lesiones solitarias pueden regresar espontáneamente (sobre todo las de localización cutánea u ósea)16. Las lesiones no son metastatizantes y el pronóstico depende del grado de afectación visceral o compresión de órganos vitales, siendo la presencia de lesiones pulmonares o cardíacas las que confieren peor pronóstico. La forma sistémica puede causar la muerte en un 73% de los casos en los que existe afectación visceral, pero este porcentaje es seguramente muy relativo al existir numerosas lesiones asintomáticas que no se diagnostican y no son tenidas en cuenta.
El tratamiento, en caso de compromiso vital o no remitir espontáneamente, es primero quirúrgico. En caso de afectación multiorgánica o lesiones de alta morbilidad podría tratarse con quimioterapia (algunos autores proponen la combinación de dactinomicina, vincristina y ciclofosfamida)7.
La recurrencia tras el tratamiento parece marcada por factores tanto clínicos (lesiones multiorgánicas, aparición en mayores de 5 años de edad, o afectación de extremidades) como histológicos (cirugía incompleta, focos de necrosis, o más de cinco mitosis por diez campos)19.
Es preciso realizar diagnóstico diferencial con dermatofibromas, leiomiomas y hemangiopericitomas20, así como con procesos más infrecuentes como el sarcoma miofibroblástico, descrito recientemente21. Este último tiene un curso agresivo, en contraste con el comportamiento generalmente benigno y autolimitado de la miofibromatosis16.


