



Las dermatosis con herencia autosómica dominante pueden presentarse en ocasiones como mosaicismos con distribución lineal o segmentaria siguiendo las líneas de Blaschko. El mosaicismo de tipo 1 está caracterizado por la presencia de un segmento corporal afectado por la enfermedad en un individuo sano, como consecuencia de una mutación germinal poscigótica en dicho segmento. El mosaicismo de tipo 2 se caracteriza por la presentación difusa de la enfermedad combinada con la sobreimposición de un segmento corporal con mayor afectación, originado por una pérdida de heterocigosidad en dicho segmento acontecida durante el desarrollo embrionario en un individuo heterocigoto para la enfermedad1.
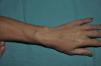
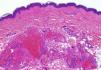
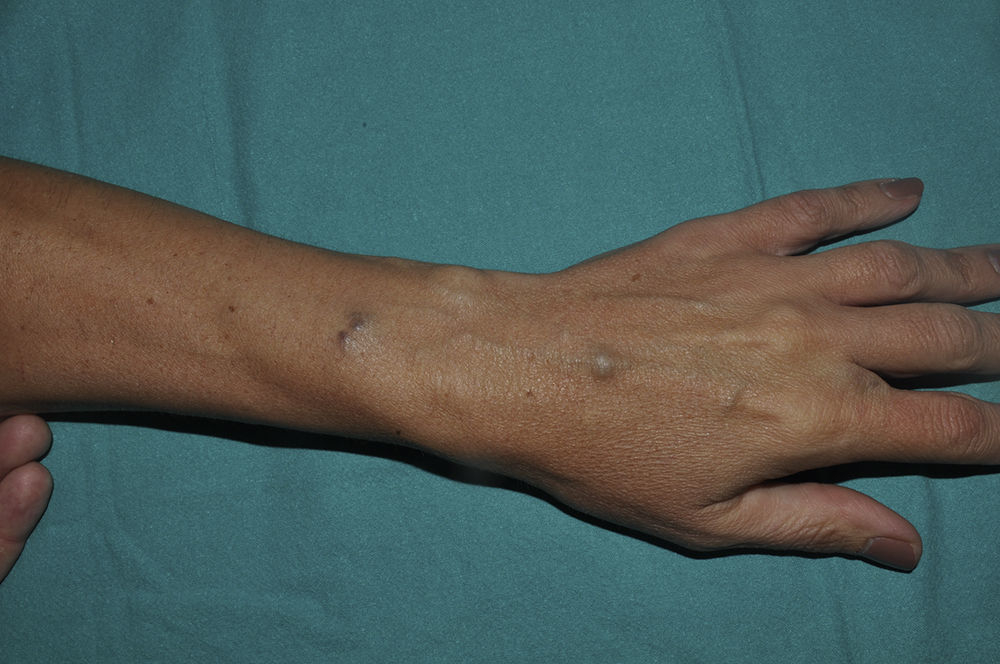
Una niña de 6 años de edad, sin antecedentes de interés, presentaba una lesión azulada congénita asintomática de distribución segmentaria, que abarcaba la ingle, la cara interna de muslo y la pierna derechas, hasta llegar al pie derecho. La lesión había crecido de forma proporcional al desarrollo corporal. Además aparecieron progresivamente desde los 3 años de edad lesiones nodulares que resultaban dolorosas a la presión y a los cambios de temperatura, que se distribuían en las 4 extremidades de forma aislada y dispersa. A la exploración se objetivó una placa azulada con nódulos palpables de consistencia elástica que se distribuían en la cara interna y posterior de muslo derecho y la cara medial del pie derecho siguiendo un trayecto segmentario (fig. 1). Además, en el pie contralateral y en zonas distales del resto de extremidades se objetivaron pápulas y nódulos azulados dispersos dolorosos a la presión. Entre los antecedentes familiares destacaba que la madre de la paciente refería desde la adolescencia la aparición salpicada de lesiones nodulares azuladas de aproximadamente 1cm de similares características a las de la paciente distribuidas en las extremidades (fig. 2) y en el tronco, que no habían sido estudiadas con anterioridad. Se decidió realizar biopsia cutánea de una de las lesiones de la madre de la paciente y la histología mostró una neoformación vascular en la dermis profunda con amplias luces vasculares rellenas de hematíes y tapizadas por células glómicas, resultando compatible con malformación glomovenosa (fig. 3). Teniendo en cuenta el antecedente de placa azulada congénita al nacimiento, la posterior aparición de lesiones dispersas y los antecedentes maternos de cuadro compatible con glomangiomatosis, realizamos el diagnóstico de mosaicismo tipo 2 en glomangiomatosis familiar. Por el momento, dada la ausencia de afectación de la calidad de vida, se ha decidido plantear una actitud expectante y seguimiento periódico de la paciente.
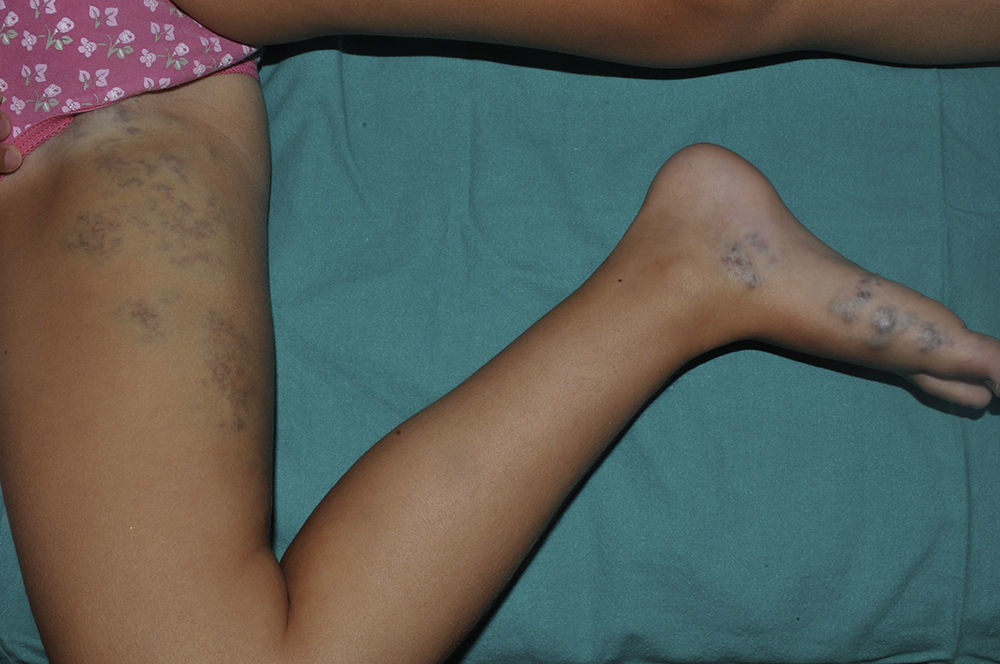
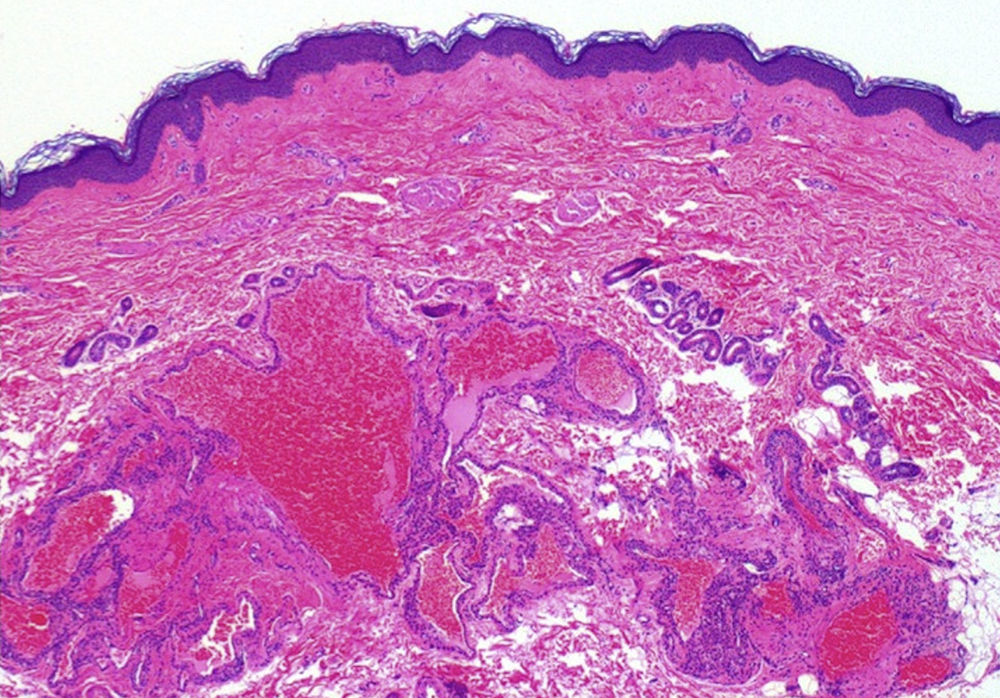
El glomangioma o malformación glomovenosa es un hamartoma derivado de los corpúsculos glómicos dérmicos, estructuras encargadas de la regulación térmica. Se describen 2 formas clínicas: el tumor glómico solitario y las malformaciones glomovenosas o glomangiomas múltiples. El primero es la forma de presentación más frecuente y se presenta típicamente en adultos como nódulos azulados, únicos y dolorosos en zonas acras. Las malformaciones glomovenosas o glomangiomas múltiples se caracterizan por la aparición progresiva a partir de la infancia o adolescencia de pápulas o nódulos azulados, aislados y dispersos, salpicados por toda la superficie corporal con herencia autosómica dominante con penetrancia incompleta o expresividad variable, por lo que solo se encuentra historia familiar previa en el 60% de los casos. Se ha demostrado que esta forma de presentación se produce como consecuencia de una mutación del gen de la glomulina, presente en el cromosoma 1p21-222. La presentación congénita de malformaciones glomovenosas es rara, y hasta el momento actual la mayoría de los casos publicados con esta forma de presentación representan un mosaicismo tipo 2, con una lesión inicial de distribución segmentaria y posterior aparición de lesiones dispersas a distancia, al igual que ocurre en el caso presentado. La glomangiomatosis familiar fue descrita por primera vez con presentación de mosaicismo tipo 2 por Happle y König3. Tras revisar la literatura hemos hallado un total de 9 casos publicados con posterioridad con este tipo de presentación clínica2–9, por lo que hasta el momento actual son 14 los casos publicados con este tipo de presentación, sin confirmación molecular de la enfermedad en ninguno de ellos. El mosaicismo tipo 2, además de en la glomangiomatosis, ha sido descrito en múltiples enfermedades caracterizadas por una herencia autosómica dominante10, aunque solo en algunas de estas enfermedades se ha confirmado el mosaicismo genéticamente.
En el caso que presentamos, a pesar de no haberse realizado estudio genético, es evidente que la madre y la niña presentan glomangiomatosis familiar. Según la presentación clínica en forma de lesión segmentaria congénita sobreimpuesta en la niña, y la posterior aparición de lesiones dispersas, consideramos que este caso podría incluirse como un nuevo ejemplo de mosaicismo tipo 2 de glomangiomatosis.


