





En las últimas décadas se ha descrito la asociación entre epidermólisis ampollosa (EA) y miocardiopatía dilatada (MD). Generalmente esta última enfermedad se detecta en fases avanzadas, implicando un peor pronóstico.
Nuestro objetivo consistió en determinar la prevalencia de MD en los pacientes con EA vistos en el Hospital San Joan de Déu (Barcelona) desde mayo de 1986 a abril de 2015.
MétodosEstudio descriptivo transversal mediante revisión de las historias clínicas con atención al tipo y subtipos mayores de EA y la existencia o no de MD.
ResultadosSe recogieron 57 pacientes con diagnóstico de EA. De ellos 19 presentaban EA simple, 10 EA juntural, 27 EA distrófica (14 EA distrófica dominante y 13 EA distrófica recesiva) y existió un caso de síndrome de Kindler. Solo 2 de los pacientes con EA distrófica recesiva presentaron MD. En 23 de los pacientes con EA existieron factores que podrían tener una relación causal con el potencial desarrollo de MD.
ConclusiónLa MD puede ser una complicación en los pacientes con EA, mayoritariamente del subtipo de EA distrófica recesiva, por lo que deben hacerse controles periódicos para su temprano diagnóstico y tratamiento.
In recent decades, an association has been reported between epidermolysis bullosa (EB) and dilated cardiomyopathy (DC). DC is typically in an advanced phase when detected, leading to a poorer prognosis. Our objective was to determine the prevalence of DC in patients with EB seen in Hospital San Joan de Déu in Barcelona, Spain, between May 1986 and April 2015.
MethodsThis was a descriptive, cross-sectional chart-review study in which we recorded the type and main subtypes of EB and the presence or absence of DC.
ResultsFifty-seven patients with EB were found, 19 with EB simplex, 10 with junctional EB, 27 with dystrophic EB (14 dominant dystrophic and 13 recessive dystrophic), and just 1 with Kindler syndrome. DC was detected in only 2 patients with recessive dystrophic EB. Twenty-three patients had presented factors that could have had a causal relationship with the potential onset of DC.
ConclusionDC is a possible complication of EB, particularly in recessive dystrophic EB. Periodic follow-up should be performed to make an early diagnosis and start treatment.
El término de miocardiopatía incluye 3 subtipos fundamentales: dilatada, hipertrófica y restrictiva. La miocardiopatía dilatada (MD) se define como una dilatación progresiva con afectación de la contractilidad del ventrículo izquierdo o de ambos1. Una característica de la MD es la insuficiencia cardíaca congestiva (ICC)2. La MD es una enfermedad infrecuente en la población pediátrica, con una incidencia de 0,6-1,2 por cada 10.000 niños1,2. Se han sugerido diversas causas de MD que incluyen formas idiopáticas, familiares o genéticas, virales, autoinmunes, por déficit de micronutrientes, por sobrecarga de hierro, por anemia crónica y etiología tóxica o farmacológica1,2.
Las epidermólisis ampollosas hereditarias (a las que nos referiremos a lo largo de todo el artículo como «epidermólisis ampollosas» [EA]) comprenden un grupo de enfermedades genéticas caracterizadas por fragilidad cutánea y formación de ampollas. Se clasifican principalmente en simple, juntural (Herlitz y no Herlitz), distrófica (dominante o recesiva) y síndrome de Kindler. La clasificación actual según el documento de consenso se muestra en la tabla 13,4.
Clasificación actual de las epidermólisis ampollosas hereditarias
| EA simples | Superficiales | Basales |
| EAS acantolítica letal Síndrome de displasia ectodérmica-fragilidad cutánea EAS superficial | EAS localizada EAS Dowling-Meara o herpetiforme EA generalizada (otras no Dowling-Meara) EAS con pigmentación moteada EAS con atresia pilórica EAS de Ogna EAS circinada migratoria | |
| EA junturales | Tipo Herlitz | Tipo no-Herlitz |
| EAJ tipo no Herlitz localizada EAJ con atresia pilórica EAJ inversa EAJ de aparición tardía Síndrome laringo-óculo-cutáneo | ||
| EA distróficas | Dominantes | Recesivas |
| EADD generalizada EADD acral EADD pretibial EADD pruriginosa EADD con distrofia ungueal solo EADD tipo dermólisis del recién nacido | EADR generalizada grave EADR generalizada (otras no graves) EADR inversa EADR pretibial EADR pruriginosa EADR centrípeta EADR tipo dermólisis del recién nacido | |
| EA mixtas | Síndrome de Kindler |
En las últimas décadas se ha observado asociación entre EA y MD, siendo el subtipo de EA distrófica recesiva el más frecuentemente asociado a miocardiopatía. Los casos publicados en la literatura indexada se muestran en la tabla 2 (la ampliación a esta se muestra en la tabla 1 del material suplementario)2,5–14.
Resumen de los casos publicados en la literatura con asociación de epidermólisis ampollosa y miocardiopatía dilatada
| Edad (años±DE) | 12,27±5,60a | |
| Sexo | V:M=27:31a | |
| FE (%) | 9–45b | |
| Tipo de EA | EAS | 3/71 (4,2%) |
| EAJ-nH | 2/71 (2,8%) | |
| EAD (no especificada) | 1/71 (1,4%) | |
| EADR | 65/71 (91,5%) | |
| Evolución | Fallecimiento | 24/71 (33,8%) |
| Supervivencia | 44/71 (62,0%) | |
| No aclarado | 3/71 (4,2%) | |
| Factores asociados citados al menos en un caso | Anemia | 22/71 (31,0%)c |
| Sobrecarga de hierro (hemosiderosis secundaria) | 2/71 (2,8%) | |
| Sobrecarga hídrica | 1/71 (1,4%) | |
| Mutación en PLEC | 1/71 (1,4%) | |
| Mutación en DSP | 2/71 (2,8%) | |
| Insuficiencia renal crónica | 6/71 (8,5%) | |
| Déficit nutricional (sin especificar) | 1/71 (1,4%) | |
| Déficit de carnitina | 7/71 (9,9%)c | |
| Déficit de selenio | 7/71 (9,9%) | |
| Déficit de cinc | 2/71 (2,8%) | |
| Hipoalbuminemia | 9/71 (12,7%) | |
| Hipoaminoacidemia | 2/71 (2,8%) | |
| Fármacos cardiotóxicos (amitriptilina, cisaprida) | 1/71 (1,4%) | |
| Etiología vírica | 1/71 (1,4%) | |
La información más detallada puede consultarse en la tabla 1 del material suplementario.
DE: desviación estándar; EA: epidermólisis ampollosa; EAD: epidermólisis ampollosa distrófica; EADR: epidermólisis ampollosa distrófica recesiva; EAJ-nH: epidermólisis ampollosa juntural no Herlitz; EAS: epidermólisis ampollosa simple; FE: fracción de eyección; M: mujer; V: varón.
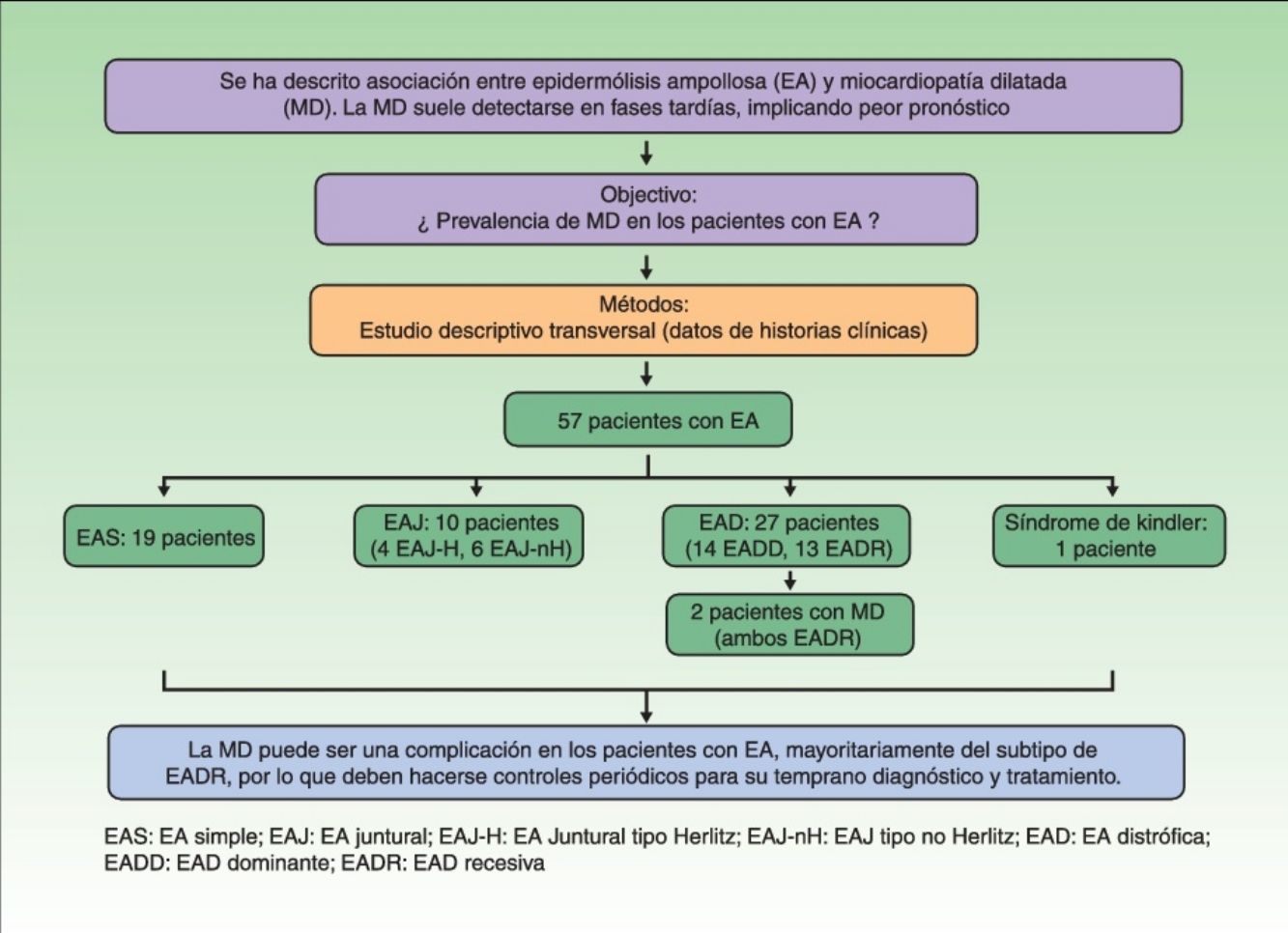
Según lo previamente expuesto, nuestro objetivo consistió en determinar la prevalencia de MD en los pacientes con EA.
MétodosSe realizó un estudio descriptivo transversal. Se recogieron todos los casos de EA vistos en el Hospital de San Joan de Déu, hospital pediátrico de referencia, entre mayo de 1986 y abril de 2015. La relación de pacientes se obtuvo a través de una base de datos en función del diagnóstico clínico, en la que se podía acceder a los números de historia clínica y fotografías de los pacientes. Los pacientes se clasificaron en los distintos tipos de EA según los datos clínicos y los estudios complementarios (histología convencional, mapeo antigénico, microscopia electrónica o estudio genético), teniendo en cuenta la clasificación del documento de consenso3. Asimismo se recogieron datos acerca del motivo de derivación a cardiología, la existencia o no de alteraciones ecocardiográficas, la presencia o no de MD (diagnosticada por un cardiólogo pediátrico, y definida como dilatación ventricular y disminución de la función sistólica) y diversos parámetros analíticos que pudieran estar en relación con la presencia de MD. La información fue recogida en todos los casos con carácter retrospectivo y basándose únicamente en lo registrado en la historia clínica (anamnesis, exploración física e informes de los estudios complementarios).
ResultadosSe obtuvieron 72 pacientes con diagnóstico de EA a través de la base de datos. Tras excluir a aquellos pacientes con diagnóstico dudoso o con pruebas complementarias incompletas para una correcta clasificación, 57 pacientes fueron finalmente incluidos en el estudio. Diecinueve presentaron EA simple, 10 EA juntural (4 EA juntural tipo Herlitz y 6 EA juntural no Herlitz), 27 EA distrófica (14 EA distrófica dominante y 13 EA distrófica recesiva) y existió un caso de síndrome de Kindler.
Se realizó valoración cardiológica y ecocardiograma en 19 pacientes (5 EA simple, uno EA juntural tipo Herlitz, 4 EA juntural no Herlitz, 2 EA distrófica dominante, 7 EA distrófica recesiva). Diecisiete pacientes no mostraban síntomas o signos de afectación cardiológica, siendo el motivo de derivación a dicho servicio la búsqueda de un diagnóstico precoz de MD. Dos pacientes fueron derivados con sospecha clínica de miocardiopatía (siendo confirmada en uno de ellos).
Solo 2 pacientes, ambos afectos de EA distrófica recesiva, presentaron MD. Esto supuso una prevalencia del 3,5% (2/57) considerando el total de los casos. La prevalencia de MD en los subtipos de EA fue del 0% en la EA simple, EA juntural y en el síndrome de Kindler (0/19, 0/10, 0/1, respectivamente), y del 7,4% (2/27) en la EA distrófica. La prevalencia de MD considerando únicamente los casos de EA distrófica recesiva fue del 15,4% (2/13).
En el primer caso la MD fue detectada a los 13 años de vida, por síntomas y signos de insuficiencia cardíaca y alteraciones ecocardiográficas compatibles (dilatación ventricular y fracción de eyección [FE]<50%). Se realizó tratamiento con carvedilol con ajuste de dosis según sintomatología y FE. Inicialmente la FE se mantuvo próxima al 50%, con marcados descensos (FE 20%) en momentos de exacerbación. Cuatro años después del diagnóstico de MD existió empeoramiento progresivo tras la aparición de insuficiencia renal secundaria a amiloidosis sistémica tipo AA. El paciente falleció a los 21 años de edad, con un importante deterioro general y fallo multiorgánico. La anemia crónica, las múltiples transfusiones periódicas, la hipoalbuminemia, la hipoaminoacidemia y el déficit de selenio y cinc podrían haber contribuido al desarrollo de MD.
En el segundo caso la paciente fue diagnosticada de MD a los 3 años, en un control ecocardiográfico rutinario. Recibe desde entonces tratamiento con carvedilol (2mg/12h), manteniendo FE en torno al 45% y ausencia de signos de insuficiencia cardíaca. Otros factores que podrían contribuir al desarrollo de MD en esta paciente incluyen anemia, transfusiones sanguíneas periódicas, hipoalbuminemia, hipoaminoacidemia y déficit de carnitina.
En la tabla 3 se muestran los factores que podrían estar en relación con un posible desarrollo de MD en los pacientes revisados, según el tipo de EA. Veintitrés pacientes presentaron alguna alteración que podría tener un papel etiológico en la MD. El 48% (11/23) de estos pacientes pertenecían al grupo de la EA distrófica recesiva.
Factores que podrían intervenir en el desarrollo de miocardiopatía dilatada en los pacientes revisados
| Factor etiológico | EAS | EAJ-H | EAJ-nH | EADD | EADR | Kindlera |
|---|---|---|---|---|---|---|
| Anemia | 10,5% (2/19) | 75% (3/4) | 66,7% (4/6) | 0% (0/14) | 76,9% (10/13) | (1/1) |
| Sobrecarga de hierro | 5,3% (1/19) | 0% (0/4) | 0% (0/6) | 0% (0/14) | 15,4% (2/13) | (0/1) |
| Déficit de selenio | 0% (0/19) | 25% (1/4) | 0% (0/6) | 0% (0/14) | 30,8% (4/13) | (0/1) |
| Déficit de carnitina | 0% (0/19) | 0% (0/4) | 0% (0/6) | 0% (0/14) | 15,4% (2/13) | (0/1) |
| Déficit de cinc | 0% (0/19) | 25% (1/4) | 0% (0/6) | 0% (0/14) | 46,2% (6/13) | (0/1) |
| Hipoalbuminemia | 5,3% (1/19) | 75% (3/4) | 16,7% (1/6) | 0% (0/14) | 69,2% (9/13) | (0/1) |
| Hipoaminoacidemia | 5,3% (1/19) | 25% (1/4) | 0% (0/6) | 7,1% (1/14) | 46,2% (6/13) | (0/1) |
| Insuficiencia renal | 0% (0/19) | 0% (0/4) | 0% (0/6) | 0% (0/14) | 7,7% (1/13) | (0/1) |
EADD: epidermólisis ampollosa distrófica dominante; EADR: epidermólisis ampollosa distrófica recesiva; EAS: epidermólisis ampollosa simple; EAJ-H: epidermólisis ampollosa juntural tipo Herlitz; EAJ-nH: epidermólisis ampollosa juntural tipo no Herlitz.
Se ha observado un mayor riesgo de cardiopatía en pacientes con EA. Fine et al. comunicaron 15 pacientes con EA e ICC. Estos autores consideraban que la mayoría de estos pacientes con ICC tenía también MD. Los subtipos de EA en los que evidenciaron cardiopatía fueron EA distrófica recesiva y EA juntural no Herlitz, siendo más frecuente en la forma de EA distrófica recesiva generalizada grave. El riesgo de ICC era significativamente mayor en estos pacientes que en la población general, y dicho riesgo se incrementaba si se asociaba insuficiencia renal crónica2. Más recientemente, Ryan et al. investigaron la presencia de daño cardíaco subclínico u otras anomalías vasculares en pacientes con el subtipo de EA distrófica recesiva. Encontraron que 40 de los 45 pacientes incluidos tenían alteraciones ecocardiográficas, y que el 18% de los casos presentaban dilatación del arco aórtico14.
En los pacientes con EA la anemia crónica, la sobrecarga de hierro, los bajos niveles de carnitina y selenio, los fármacos y las infecciones virales concomitantes son los factores que más se han relacionado con el desarrollo de cardiopatía1.
La anemia crónica es generalmente de etiología multifactorial, con pobre respuesta a suplementos dietéticos y con requerimiento habitual de transfusiones periódicas. Tanto la anemia crónica como la sobrecarga de hierro ocasionada por repetidas trasfusiones pueden desempeñar algún papel en el desarrollo de la MD1.
En cuanto a los déficits nutricionales se ha documentado una diferencia estadísticamente significativa entre los niveles de carnitina de pacientes con EA y MD con respecto a pacientes con EA sin MD, siendo menores en el primer grupo1. Se ha observado que los suplementos de selenio en estados carenciales reducen la morbimortalidad en casos de MD. Sin embargo, no se encontraron niveles de selenio significativamente inferiores en pacientes con EA y MD en comparación con los pacientes con EA sin MD8. El déficit de tiamina es también una causa documentada de MD8. Aunque se han descrito casos de reversibilidad de MD con reposición de los niveles de carnitina8, o mejoría de la sintomatología con suplementos de selenio o de tiamina, los niveles de un micronutriente disminuido de modo aislado no parecen ser un único factor causal de MD, pero podrían contribuir a su desarrollo1,13.
Las infecciones víricas pueden provocar MD1. Habitualmente el daño cardíaco se debe a un proceso autoinmune postinfeccioso8. Muchas veces el cuadro viral prodrómico pasa desapercibido porque puede preceder al desarrollo de la MD en varios meses. El diagnóstico es incluso más difícil en pacientes con formas agresivas de EA, en los que pueden aparecer síntomas como fiebre por diversos motivos o disnea de origen no cardíaco1.
Los fármacos cardiotóxicos pueden exacerbar la MD en estos pacientes, por lo que deberían ser suspendidos en caso de sintomatología sugestiva de cardiopatía15.
Hasta el momento no se ha documentado la existencia de otras mutaciones genéticas que afecten a proteínas de la membrana basal que lleven al desarrollo de la miocardiopatía1,8, salvo en los casos de EA simple con distrofia muscular asociada a MD por mutaciones del gen PLEC1 (que codifica para la plectina)12,16,17, y de EA simple acantolítica letal por mutaciones en el gen DSP (que codifica para la desmoplaquina)18,19. De manera interesante se ha descrito un paciente con EA distrófica recesiva con miocardiopatía no compactada del ventrículo izquierdo (MNVI) sin MD20. Los autores de este artículo indican que en algunos casos la MNVI evoluciona a MD, sugiriendo que la MNVI podría preceder a la MD en las EA distróficas recesivas.
Además de este riesgo incrementado para MD, los pacientes con EA y MD tienen una mortalidad de un 30-60%. En el subtipo de EA distrófica recesiva la MD suele detectarse a los 10 años de edad, en una fase avanzada, con una FE baja y una mortalidad elevada, que generalmente ocurre alrededor de los 3 meses después del diagnóstico. Estos datos sugieren un retraso en el diagnóstico de la MD, implicando un difícil tratamiento1.
En cuanto al manejo diagnóstico, se ha propuesto determinación analítica con hemograma, albúmina, carnitina total y libre, selenio y cinc y evaluación cardiológica (con electrocardiograma y ecocardiograma) con periodicidad anual en los pacientes asintomáticos. En aquellos sintomáticos o con algún parámetro alterado en las pruebas previamente citadas, se aconseja valoración cardiológica urgente, determinación de hormonas tiroideas, serologías, cultivo de parvovirus, Coxsackie virus, determinaciones metabólicas y biopsia de miocardio si procede1.
ConclusionesEs importante tener en cuenta la MD como una posible complicación de la EA, sobre todo en los tipos de EA distrófica recesiva y EA juntural. Por tanto, deben hacerse evaluaciones precoces y periódicas, así como corrección subsiguiente de los factores que se han relacionado con el daño cardíaco en estos pacientes1. Una detección temprana de dichas alteraciones permitirá una pronta instauración del tratamiento necesario, lo que retrasará la progresión clínica y prolongará la supervivencia en los pacientes con EA.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.



