




INTRODUCCION
Bajo la denominación de micosis fungoide (MF) se engloban muy diversas lesiones cutáneas con diferentes matices clínicos e histológicos1. Uno de ellos es la presencia de granulomas, lo cual define la micosis fungoide granulomatosa (MFG), una rara forma de MF de la que existen escasos casos publicados2, 3 desde su descripción por Ackerman en 19704.
Dada la poca frecuencia de esta variante de MF, presentamos aquí un caso de MFG con las particularidades de ser una lesión única de más de 10 años de evolución.
DESCRIPCION DEL CASO
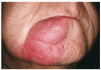
Se trata de una mujer de 68 años sin antecedentes de interés y sin ingesta habitual de medicación. En 1995 consultó por presentar lesión asintomática mandibular de más de 5 años de evolución. En la exploración presentaba en lado derecho del mentón una placa eritematoedematosa de 5 × 5 cm, con bordes netos, superficie lisa e infiltrada al tacto (fig. 1). El resto de la exploración cutánea era normal.
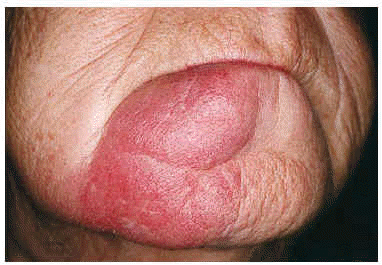
Fig. 1. Placa eritematoedematosa de consistencia blanda en mentón. Lesión asintomática de más de 10 años de evolución.
Esta lesión había sido biopsiada en cuatro ocasiones con similares hallazgos histológicos. El principal hallazgo era la presencia en toda la dermis de un denso infiltrado inflamatorio con granulomas constituidos por histiocitos epitelioides y células gigantes multinucleadas en su parte más central y linfocitos en periferia; en varios de ellos existían áreas de caseosis (figs. 2 y 3). Los linfocitos del infiltrado eran monomorfos sin atipias. Se detectaron focos aislados de epidermotropismo, sin franca atipia nuclear en los linfocitos; no se evidenciaron microabcesos de Pautrier; en algunas áreas existía fibrosis laminar en dermis papilar; en algunos cortes se evidenciaron áreas de mucinosis folicular; algunas células gigantes presentaban signos de linfofagocitosis. En el estudio inmuhistoquímico destacaba la expresión de CD3 y CD4 en el 80% del infiltrado linfocitario, del CD68 en las células epiteloides del centro de los granulomas y de la S100 en células dispersas dentro del infiltrado inflamatorio (no en células gigantes). No se observaron signos de elastólisis con la tinción de Van Gieson, ni cuerpos extraños con la luz polarizada, ni microorganismos con las tinciones de Ziehl-Neelsen y PAS. La visión directa de leishmanias, el cultivo para microbacterias y la PCR para Mycobacterium avium intracelulare, Mycobacterium tuberculosis y Mycobacterium leprae fueron negativas. No pudo realizarse estudio de reordenamiento genético.
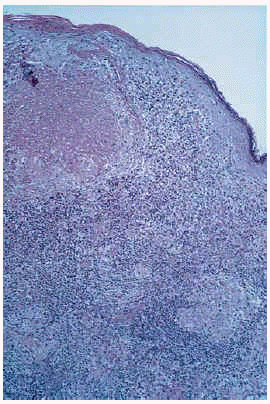
Fig. 2. Infiltrado en dermis superficial y profunda con formación de granulomas; algunos de los granulomas presentan áreas de necrosis.
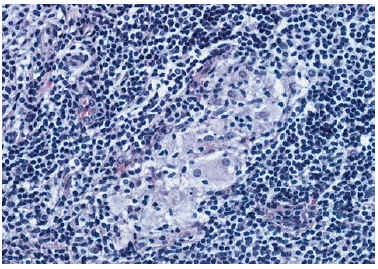
Fig. 3. Detalle de los granulomas compuestos por linfocitos, células epiteliodes y células gigantes.
Ninguna de las siguientes pruebas complementarias mostró datos relevantes: hemograma, bioquímica, proteinograma, calcemia-calciuria, inmunoglobulinas, auto-AC, serologías (lúes, virus de la inmunodeficiencia humana), enzima conversora de la angiotensina (ECA), proteína de Bence-Jones, radiografía de tórax y ortopantomografía. Se habían realizado tratamientos sistémicos previos con corticosteroides, tetracilinas, penicilina y claritromicina, así como diversas terapéuticas tópicas sin mejoría alguna. Tras descartar la sarcoidosis y la infección por micobacterias se estableció el diagnóstico de MFG.
Durante los 6 años de control en nuestra consulta (mínima evolución de 12 años), la placa ha permanecido estable y no se han detectado otras lesiones cutáneas, adenopatías, ni alteraciones sistémicas. Dada la ausencia de sintomatología y la nula repercusión sistémica, la paciente se ha opuesto reiteradamente a la realización de los tratamientos locales propuestos (radioterapia o quimioterapia con mostaza nitrogenada).
DISCUSION
Desde el punto de vista clínico, la MFG puede presentarse con una clínica muy variada, tal y como se refleja en las dos series más recientes que recopilan los casos publicados2, 3. La mayoría de las MFG se han manifestado en forma de pápulo-placas eritematosas, y no tenemos conocimiento de MFG unilesionales, salvo un caso (comunicación oral) del Hospital de La Princesa5.
Existe clara controversia en la bibliografía respecto a si la MFG presenta un mejor o peor pronóstico que otras formas de MF. Varios artículos muestran su asociación con una rápida afectación extracutánea, lo cual conlleva un mal pronóstico6, 7. Por otra parte, la presencia de granulomas se considera reflejo de una buena respuesta inmunológica frente a antígenos tumorales, y así parecen avalarlo casos con evolución crónica exclusivamente cutánea e incluso con resolución espontánea2, 4, 8. Nuestro caso se englobaría dentro de las MF unilesionales, las cuales conllevan una evolución crónica y benigna. En este sentido hemos encontrado una única referencia bibliográfica de una «hipotética» variedad de MF, la «MFG juvenil», que aparecería en edades tempranas, con clínica muy diversa, histología sarcoidea y evolución indolente durante años9.
Salvo en la enfermedad de Hodgkin, la presencia de granulomas en procesos linfoproliferativos es rara, pero se ha descrito en muy diversos linfomas cutáneos, incluso en linfomas B con epidermotropismo10. Como dato genérico se estima que un 4% de las MF pueden presentar granulomas en el estudio histológico. Cuando no existen signos histológicos inequívocos de MF, la presencia de granulomas complica y/o retrasa el diagnóstico de MFG. De hecho, un porcentaje importante de estos casos se etiquetan inicialmente de sarcoidosis11, 12, sobre todo por esta ausencia de alteraciones histológicas específicas de MF. La mayoría de los granulomas en la MFG son linfohistiocitarios con células gigantes, pero también se han descrito granulomas sarcoideos y con necrobiosis13. En nuestro caso existían granulomas de estas tres variantes señaladas.
Por esta constante existencia de granulomas se ha relacionado la MFG con el síndrome de la piel laxa granulomatosa. Esta segunda entidad se presenta clínicamente con áreas de piel péndula en las flexuras y desde el punto de vista histológico las células gigantes, con signos de fagoelastólisis, son el hallazgo más relevante14. No obstante, a pesar de estas teóricas diferencias, son entidades que pueden superponerse15.
En resumen, con este caso hemos querido, en primer lugar, recordar la dificultad del diagnóstico clínico-histológico de la MFG y considerar la sospecha de este cuadro ante infiltrados granulomatosos persistentes que no reúnen criterios diagnósticos de otras dermatosis granulomatosas. En segundo lugar hemos pretendido aportar un dato más a la ya compleja expresividad clínica de la MF, en este caso un paciente con una única lesión de MFG de evolución benigna a lo largo de 10 años.


