


INTRODUCCIÓN
La micosis fungoide es la forma más frecuente de linfoma cutáneo de células T. Las manifestaciones cutáneas típicas incluyen las fases de mancha, placa, tumor y eritrodermia, aunque se describen múltiples variantes clinicomorfológicas atípicas, incluyendo formas pustulosas, ampollosas, hiperpigmentadas, purpúricas, foliculares y verrugosas, entre otras1,2.
Una de las formas menos frecuente de presentación es la denominada micosis fungoide acrómica, que se ha descrito casi exclusivamente en pacientes de piel oscura, fundamentalmente asiáticos, y sobre todo en niños de edades entre los 2 y los 19 años. Desde la descripción inicial por Ryan et al en 19732,3, se han publicado 113 casos de pacientes de los que solamente 16 son de raza blanca2,4-10.
DESCRIPCIÓN DEL CASO
Un varón de 23 años acudió al servicio de dermatología por la presencia de unas lesiones asintomáticas en los muslos, de unos 3 años de evolución, que sufrían brotes de agudización, con extensión centrípeta hacia los glúteos y la región abdominal, que mejoraban parcialmente sin tratamiento. El paciente tenía antecedentes de neumonía por aspiración en el nacimiento, convulsiones febriles en la infancia, rinoconjuntivitis alérgica y toma ocasional de alprazolam. Entre los antecedentes familiares de interés, cabe destacar la intolerancia materna al níquel.
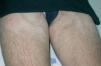
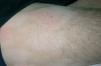
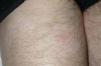
A la exploración, en las caras anterior y posterior de la raíz de los muslos, y extendiéndose hacia la zona suprapúbica, se observaban múltiples placas acrómicas, algunas con superficie eritematosa, finamente descamativa, con bordes hipocrómicos tenues, no infiltradas al tacto y confluentes en placas de mayor tamaño, algunas del tamaño de la palma (figs. 1-3). No se palpaban visceromegalias o masas abdominales, ni tampoco adenopatías locorregionales. El tratamiento con corticoides tópicos de baja potencia y emolientes indujo una mejoría con posterior reaparición de las lesiones en un período de 3 o 4 meses.
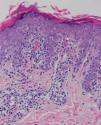
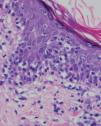
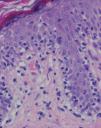
Se practicaron varias biopsias cutáneas, algunas de ellas sin alteraciones significativas. En otras se observó (figs. 4-6) una epidermis con hiperqueratosis y paraqueratosis focal, elongación de las crestas interpapilares y focos de degeneración hidrópica basal, con algún cuerpo apoptótico aislado. En dermis superficial y media existía un infiltrado linfocitario perivascular. Llamaba la atención la exocitosis de linfocitos atípicos intraepidérmicos, alguno de ellos con halo claro perinuclear y en alguna zona incluso iniciando la formación de microabscesos de Pautrier.
Ante la conjunción de los hallazgos clínicos e histopatológicos descritos, se estableció el diagnóstico de micosis fungoide, en su variante acrómica, y se solicitó una serie de pruebas complementarias (hemograma, bioquímica, coagulación, orina, hormonas tiroideas y radiografía de tórax) que fue rigurosamente normal. Los estudios de inmunohistoquímica para determinar el inmunofenotipo demostraron la naturaleza T del infiltrado linfocitario (CD3+), compuesto fundamentalmente de células supresoras/citotóxicas (CD8+), con un claro predominio sobre los linfocitos CD4. El estudio de reordenamiento genético no mostró clonalidad.
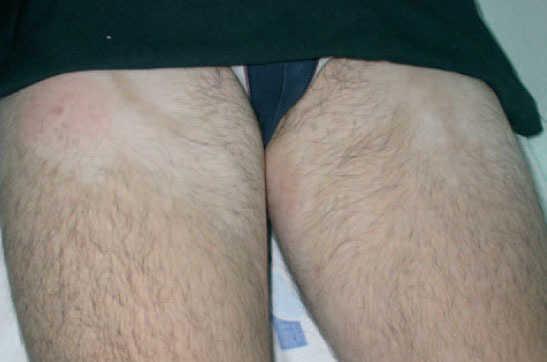
Fig. 1.—Manchas acrómicas en tercio proximal de muslos.
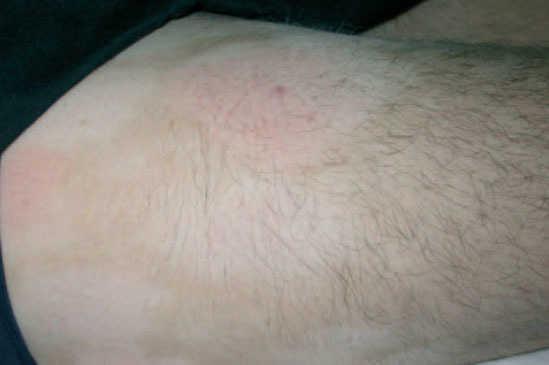
Fig. 2.—Manchas acrómicas de centro eritematoso descamativo.
El paciente comenzó tratamiento con corticoides tópicos de mediana potencia y experimentó una mejoría parcial. Actualmente se encuentra asintomático y no ha presentado nuevas lesiones ni ninguna otra manifestación sistémica, aunque se le realizan revisiones periódicas para controlar su evolución y está pendiente de comenzar tratamiento con psoraleno y luz ultra-violeta (PUVA), en caso de que se produzca un empeoramiento.
DISCUSIÓN
La frecuencia real de la micosis fungoide hipopigmentada es aún desconocida y posiblemente esté infradiagnosticada, ya que la mayoría de los pacientes descritos hasta la actualidad tenía diagnósticos clínicos previos de vitíligo, dermatitis atópica, pitiriasis liquenoide crónica, hipopigmentación postinflamatoria, lepra, tiña versicolor, pitiriasis alba y sarcoidosis, entre otros2,11. Con frecuencia, el diagnóstico definitivo mediante estudio histológico se retrasa una media de 2 a 12 años6,12-20, por el curso lento, progresivo e indolente de la enfermedad.
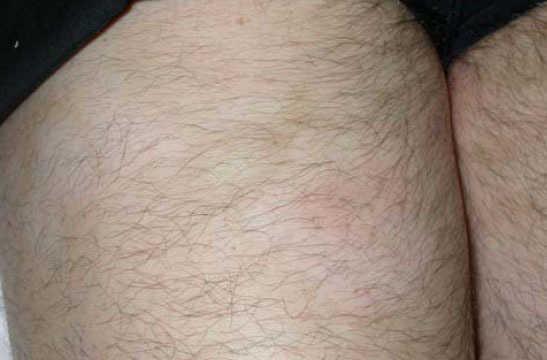
Fig. 3.—Imagen de detalle de las lesiones.
La forma más habitual de presentación clínica consiste en grandes máculas hipopigmentadas en el tronco y en las extremidades, habitualmente asintomáticas, aunque se puede asociar un prurito moderado11,14-16. En ocasiones, las manchas aparecen perfectamente delimitadas2, lo cual facilita el diagnóstico; sin embargo, en muchos casos los bordes de las lesiones aparecen muy poco definidos, lo cual dificulta la sospecha diagnóstica, casos en los que se plantean los diagnósticos diferenciales previamente citados. Las lesiones hipopigmentadas de la micosis fungoide pueden ser la única manifestación de la enfermedad, o bien se pueden asociar con otras manchas, placas o tumores. En pacientes de raza blanca se describe un patrón mixto, con componente eritematoso sobreañadido a las placas acrómicas. Probablemente, en pacientes de piel oscura, en los que esta forma es más frecuente, el componente eritematoso también está presente, pero su identificación resulta más difícil.
Como ocurrió con nuestro paciente, el estudio histopatológico en los estadios iniciales de la enfermedad puede ser muy inespecífico, lo que motiva además el retraso diagnóstico. En las fases más avanzadas revela la presencia de epidermotropismo de linfocitos anómalos, que a veces constituyen microabscesos de 5,6,8,11-23. En contraste con la micosis fungoide clásica, en esta variante parece que se observa un epidermotropismo más llamativo en contraste con un infiltrado linfocitario moderado en la dermis24.
El paciente con micosis fungoide acrómica a menudo muestra un fenotipo de linfocitos T supresores (CD8+), al contrario de lo que sucede con la forma clásica de micosis fungoide, que suele presentar un predominio de positividad para CD42,4.
Se ha señalado que la positividad para CD8 podría ser característica de la micosis fungoide hipopigmentada en niños y pacientes de piel oscura2.
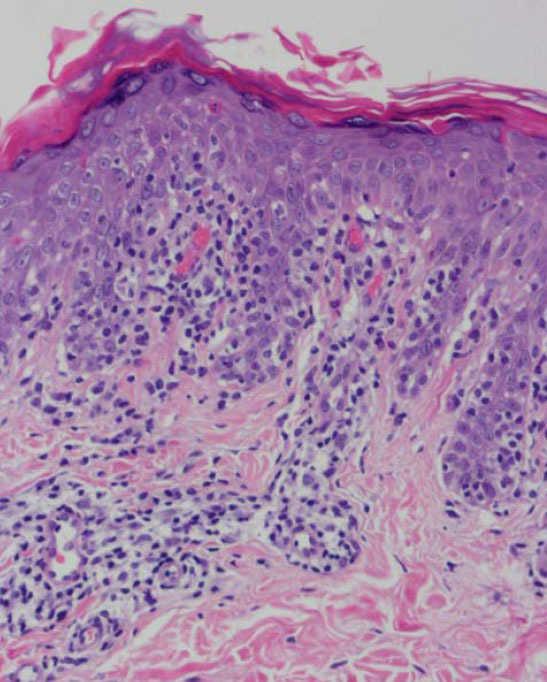
Fig. 4.—Paraqueratosis focal, elongación de crestas interpapilares, focos de degeneración hidrópica basal. (Hematoxilina-eosina, ×40.)
La patogenia no está clara2,25, de forma que se plantean diversas hipótesis. Así, se cree que el efecto citotóxico de los linfocitos T supresores sobre los melanocitos causaría la degeneración de éstos con alteración de la melanogénesis, originando las placas acrómicas, al menos en los casos con inmunofenotipo CD8+. Algunos autores no encuentran bloqueo en la transferencia de los melanosomas desde los melanocitos a los queratinocitos12,24,26, mientras que otros sugieren que este bloqueo sería el origen de las lesiones acró-micas11-14.
El diagnóstico de la micosis fungoide acrómica se basa en una buena correlación clinicopatológica, pues el análisis de inmunohistoquímica puede poner de manifiesto subpoblaciones linfocitarias T, tanto CD4 como CD8+.
El estudio de reordenamiento genético, en la mayoría de los casos, al igual que en el nuestro, no demuestra monoclonalidad. En cualquier caso, la monoclonalidad no es sinónimo de malignidad, pues numerosas dermatosis benignas, incluyendo casos de vitíligo inflamatorio, pueden presentar una proliferación monoclonal de linfocitos T2,27. El tratamiento de la variante acrómica no difiere apenas de la forma clásica de micosis fungoide; la terapia con PUVA induce remisiones completas y rápidas en la mayoría de los casos y puede considerarse la primera línea de tratamiento7,12,13,16,18,24. También han resultado útiles la radiación ultravioleta B (UVB), la mostaza nitrogenada, la carmustina tó-13,24 y los corticoides tópicos potentes24,28 en estadios iniciales de la enfermedad25. Incluso sin tratamiento, ésta parece seguir un curso benigno17. En ocasiones pueden producirse recidivas que responden rápidamente a un nuevo ciclo de tratamiento25. El seguimiento de los pacientes con este tipo de micosis fungoide acrómica demuestra que siguen un curso similar al de la micosis fungoide típica en estadio Ia. Aunque existen casos de progresión hacia la enfermedad sistémica y la muerte del paciente26, lo más habitual es que el curso del proceso no sea agresivo y no exista afectación ganglionar ni sistémica4,26.
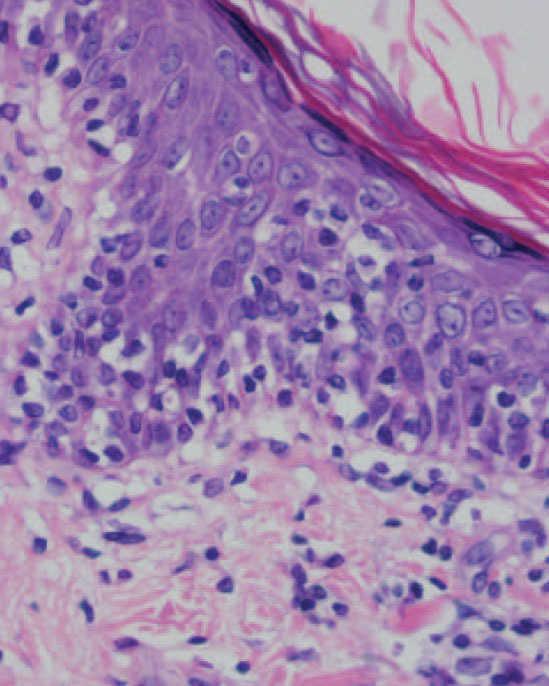
Fig. 5.—Exocitosis intraepidérmica de linfocitos atípicos. (Hematoxi-lina-eosina, ×100.)
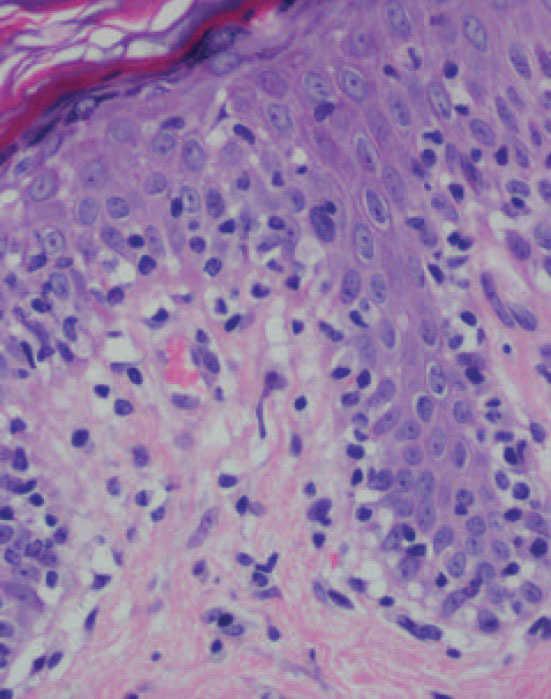
Fig. 6.—Exocitosis linfocitaria que muestra degeneración hidrópica basal. (Hematoxilina-eosina, ×100.)
En los casos con inmunofenotipo CD8+, los resultados de la inmunohistoquímica deben ser analizados con precaución. Cribier et al27 describieron 4 casos de vitíligo que simulaban una micosis fungoide. A pesar de la existencia de linfocitos hipercromáticos con exocitosis, estos casos se diagnosticaron como vitíligo por el predominio de CD8+ epidermotropos y la ausencia de melanocitos27. Posiblemente sean precisos estudios moleculares adicionales para diferenciar con precisión estos casos de micosis fungoide acrómica27,29.
El diagnóstico diferencial de manchas hipopigmentadas en pacientes jóvenes, no sólo de piel oscura, sino también en pacientes de raza blanca debe incluir la variante acrómica de la micosis fungoide. Ante lesiones de estas características y refractarias a los tratamientos, debe realizarse siempre un estudio histopatológico, precisándose en ocasiones múltiples biopsias para llegar al diagnóstico definitivo.
Correspondencia:
Pablo de Unamuno. Cuesta de Sancti Spiritus, 6, 3.º B.
37001 Salamanca. España.
unamunop@usal.es
Recibido el 3 de marzo de 2004.
Aceptado el 9 de junio de 2004.


