


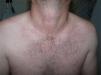
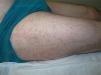

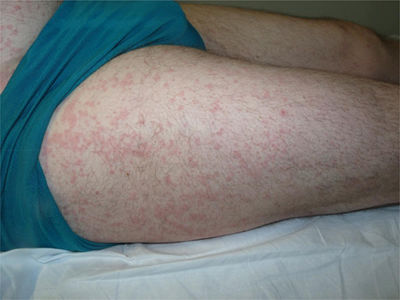
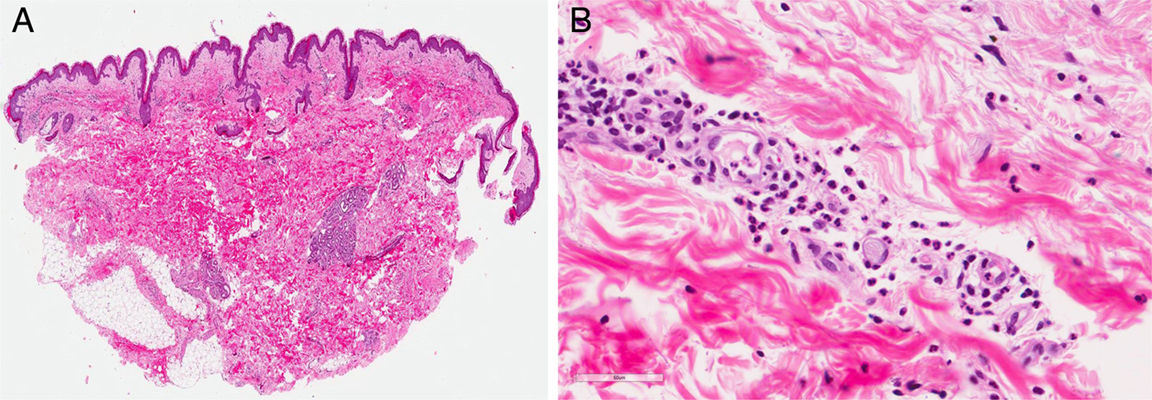
Un varón de 44 años de edad es valorado por una erupción cutánea de 7 días de evolución asociado a fiebre vespertina y dolores articulares. Refería un cuadro catarral una semana previa con dolor faríngeo. Trabajaba de ganadero y no refería antecedentes de contactos sexuales de riesgo ni viajes recientes. A la exploración física se objetivó una erupción maculopapular color «rosa salmón» localizada principalmente en el tronco (fig. 1) y partes proximales de miembros superiores e inferiores (fig. 2), sin afectación de palmas ni plantas. Además, presentaba fiebre >39°C y artritis en articulaciones de hombro derecho, rodilla y tobillo izquierdos. El resto de la exploración constató adenopatías axilares e inguinales sin hepatoesplenomegalia. Se realizó una biopsia cutánea que evidenció una epidermis normal con un infiltrado linfohistiocitario perivascular superficial en dermis acompañado de neutrófilos, sin otro tipo de alteración (fig. 3).
Pruebas complementarias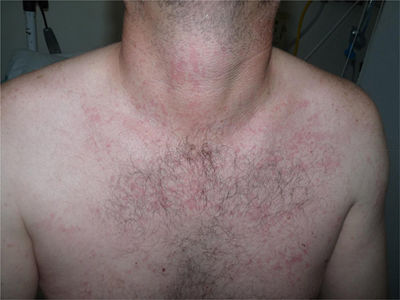
Hemograma con leucocitos: 13.300leu/μl (Seg 83,7%), velocidad de sedimentación globular: 15mm; bioquímica con GOT: 22UI/l, GPT: 20UI/l, ferritina: 1.148ng/ml, proteína C reactiva: 13,7mg/dl. Los anticuerpos anti-nucleares (ANA), el factor reumatoide (FR), las serologías microbianas, hemocultivos y urocultivos fueron negativos. Las cifras del complemento, inmunoglobulinas y el proteinograma fueron normales.
¿Cuál es su diagnóstico?DiagnósticoEnfermedad de Still de presentación en el adulto.
Evolución y tratamientoEl paciente cumplía criterios diagnósticos de enfermedad de Still de presentación en el adulto (ES-A)1. Se pautó naproxeno 500mg/12h y prednisona 20mg/día en pauta descendente, con mejoría, pero con nuevos rebrotes al bajar las dosis de corticoides, por lo que se añadió metotrexato 10mg/semanal. Actualmente se encuentra con dosis de 5mg/día de prednisona y 2,5mg/semana de metotrexato, con buen control clínico al año de seguimiento.
ComentarioLa ES-A es una entidad infrecuente1, descrita en 1971 por Bywaters, y clasificada recientemente como un trastorno auto-inflamatorio poligénico2. Se presenta generalmente entre la tercera y sexta década, con predominancia en mujeres. La fisiopatología y los factores desencadenantes continúa sin aclarar2. Se han propuesto varias teorías3, incluyendo infecciones, que actuarían como «activadores», principalmente virales; se han relacionado con marcadores genéticos (HLA DRB1*1201, 1501, B35, DR2, DR5)3. Clínicamente muestra picos febriles vespertinos, junto a una erupción maculopapular evanescente de color «rosa-salmón», artritis, linfadenopatías y/o hepatoesplenomegalia. Dentro de los diagnósticos diferenciales se incluyen la urticaria, las vasculitis, las toxicodermias, el síndrome de Sweet, las neoplasias hematológicas, etc. En la histopatología es característico un infiltrado linfohistiocitario perivascular en dermis con neutrófilos. En la analítica destaca leucocitosis, hiperferritinemia y elevación de enzimas hepáticas. Los ANA y el FR son negativos3,4. Además, pueden evidenciarse niveles elevados de IL-6, TNF-alfa, IFN-γ, IL-183. Los niveles de ferritina, aunque con baja sensibilidad y especificidad, son generalmente superiores a 1.000ng/ml, y se correlacionan con la actividad de la enfermedad y usualmente se normalizan con la remisión de la misma3; incluso podrían predecir una progresión crónica5. La ferritina glucosilada presenta mayor especificidad. Se ha propuesto, que la expresión de la proteína quinasa MAP4K3 se correlaciona con la actividad de la enfermedad y está involucrada en la patogénesis6.
De acuerdo al curso de la enfermedad y el perfil de citoquinas que predominan, puede ser dividida principalmente en 2 fenotipos: ES-A de patrón sistémico (PS) y ES-A patrón articular (PA)2. LA ES-A puede presentar diversas complicaciones4, aunque son raras, e incluyen: hepatitis, pleuritis, neumonitis y pericarditis; dentro de las complicaciones graves se encuentran además el síndrome de activación macrofágica (SAM) y la coagulación intravascular diseminada (CID), ambas con elevado riesgo de mortalidad. En un estudio estas complicaciones se presentaron en un 14,8% de los casos1, siendo el sexo femenino el asociado a mayor riesgo1,3. La presencia de pancitopenia y síndrome de distrés respiratorio deberían alertar la posibilidad de SAM3.
El tratamiento de la ES-A se basa en la experiencia de casos individuales y/o series de casos2, entre los fármacos utilizados se encuentran antiinflamatorios no esteroideos, corticoides, metotrexato, hidroxicloroquina, leflunomida, azatioprina, ciclosporina, penicilamina o tacrolimus. También se ha empleado fármacos biológicos en casos graves y refractarios como infliximab, etanercept, adalimumab, anakinra y tocilizumab.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


