



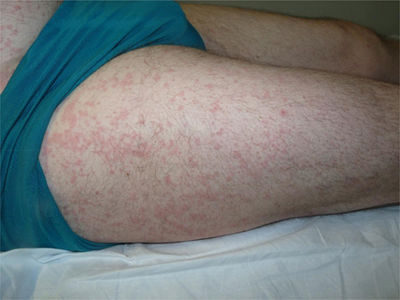
A 44-year-old man was assessed for a skin rash associated with evening fever and joint pain that had appeared 7 days earlier. He reported catarrhal symptoms and pharyngeal pain in the previous week. He worked as a livestock farmer and stated that he had not traveled recently or any risky sexual behavior. Physical examination showed a salmon-colored maculopapular rash mainly affecting the trunk (Fig. 1) and the proximal areas of the upper and lower limbs (Fig. 2), with sparing of the palms and soles. The patient also had fever (>39°C) and arthritis affecting the joints of his right shoulder and left knee and ankle. No enlargement was noticed in the axillary or inguinal lymph nodes or in the liver or spleen. Skin biopsy showed a normal epidermis with a superficial perivascular lymphohistiocytic dermal infiltrate with neutrophils. No other abnormal findings were observed (Fig. 3).
Additional Tests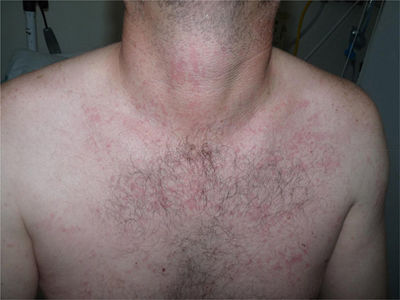

Complete blood count with leukocytes, 13300leukocytes/μL (segment, 83.7%); erythrocyte sedimentation rate, 15mm; biochemistry with aspartate aminotransferase, 22IU/L; alanine aminotransferase, 20IU/L; ferritin, 1148ng/mL; and C-reactive protein, 13.7mg/dL. Antinuclear antibodies, rheumatoid factor, microbial serology, and blood and urine cultures were all negative. Complement, immunoglobulin, and protein levels were normal.
What Is Your diagnosis?DiagnosisAdult-onset Still disease.
Clinical Course and TreatmentThe findings were compatible with a diagnosis of adult-onset Still disease (AOSD).1 The patient was started on naproxen 500mg/12h and a tapering course of prednisone 20mg/d. He showed initial improvement, but the disease flared up again when the corticosteroid doses were reduced. Methotrexate 10mg/wk was added to the treatment. At the time of writing, after a year of follow-up, the patient has achieved good clinical control of the disease with prednisone 5mg/d and methotrexate 2.5mg/wk.
CommentAOSD is an uncommon disease described by Bywaters in 1971.1 It was recently classified as a polygenic autoinflammatory disease.2 It generally appears between the third and sixth decade of life and is more common in women. Its pathophysiology and triggers remain unclear,2 although several theories have been proposed,3 including infections, which would largely act as viral “activators”. A number of genetic markers (HLA DRB1*1201, 1501, B35, DR2, DR5) have also been implicated.3 Clinically, the disease is characterized by a fever that spikes in the evening, an evanescent salmon-colored maculopapular rash, arthritis, enlarged lymph nodes, and/or hepatosplenomegaly. The differential diagnosis should include numerous entities such as urticaria, vasculitis, drug eruptions, Sweet syndrome, and hematologic tumors. The hallmark histopathologic finding is a perivascular lymphohistiocytic dermal infiltrate with neutrophils. Remarkable findings in the blood tests are leukocytosis, hyperferritinemia, and elevated liver enzymes. Antinuclear antibodies and rheumatoid factor are negative.3,4 There may also be high levels of interleukin (IL) 6, tumor necrosis factor, interferon-γ, and IL-8.3 Although ferritin levels have low specificity and specificity, they generally exceed 1000ng/mL and correlate with disease activity. They typically return to normal on remission of the disease3 and they may even predict a chronic course.5 Glycosylated ferritin is a more specific marker. Levels of the kinase protein MAP4K3 might be correlated with disease activity and may even have a pathogenic role6
AOSD can be divided into 2 main phenotypes, systemic-onset AOSD and articular AOSD, based on disease course and cytokine profile.2 Complications are rare4 but can include hepatitis, pleuritis, pneumonitis, and carditis. More serious complications are macrophage activation syndrome and disseminated intravascular coagulation, both of which have a high mortality. In 1 series, 14.8% of patients developed severe complications1 and female sex was the greatest risk factor.1,3 Pancytopenia and respiratory distress syndrome should raise suspicion of the possibility of macrophage activation syndrome.
The treatment for OASD is based on experiences with individual patients and/or small series.2 Pharmacologic treatments include nonsteroidal anti-inflammatory drugs, corticosteroids, methotrexate, hydroxychloroquine, leflunomide, azathioprine, ciclosporin, penicillamine, and tacrolimus. Biologic drugs have also been used in severe, refractory cases and include infliximab, etanercept, adalimumab, anakinra, and tocilizumab.
Conflicts of InterestThe authors declare that they have no conflicts of interest.
Please cite this article as: Flores-Terry MÁ, Franco-Muñóz M, Garrido-Martín JA, Villasanti-Rivas N. Máculas evanescentes en tronco y extremidades. Actas Dermosifiliogr. 2018;109:645–646.


