
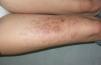
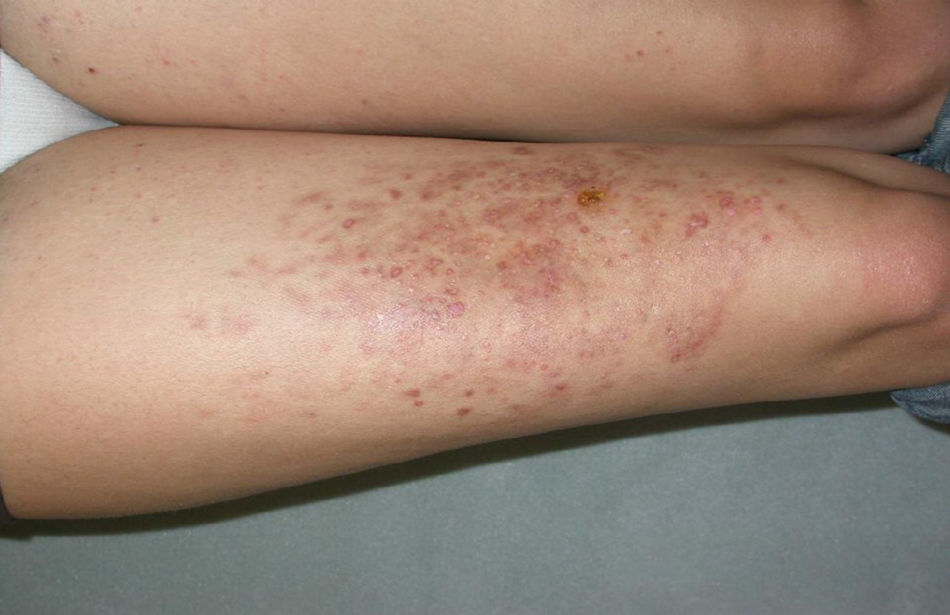
El dermatofibroma (DF) es el tumor fibrohistiocitario más frecuente de la piel y una de las neoplasias benignas que origina mayor número de consultas en la práctica clínica dermatológica. Suele presentarse como una lesión aislada, siendo más infrecuente la presentación en forma de lesiones múltiples. El término «DF múltiples» define la aparición de al menos 15 dermatofibromas en pocos meses1 o de 5-8 dermatofibromas en 4 meses2. Existe además una forma de presentación como lesiones múltiples agrupadas mucho más infrecuente: «DT múltiples agrupados», descrita por primera vez en el año 1984 por Dupré et al3. Presentamos un nuevo caso de esta rara entidad. Se trata de una mujer de 25 años de edad que acudió a consulta por lesiones papulosas de pequeño tamaño, eritematosas, con centro más oscuro y periferia más clara y máculas hiperpigmentadas residuales en menor número, asintomáticas, en la cara anterior del muslo derecho, que aparecieron cuando tenía 1 año de edad, aumentaron en número y tamaño en 2-3 meses y, desde entonces, se han mantenido estables, con remisión parcial de alguna lesión de forma aislada (fig. 1). La paciente no refería antecedentes generales ni locales de interés. Aportaba una resonancia magnética nuclear de muslo, que se había realizado previamente con el fin de descartar linfangioma, en la que no se observaba atrofia grasa o muscular ni otra alteración relevante.
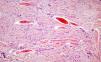
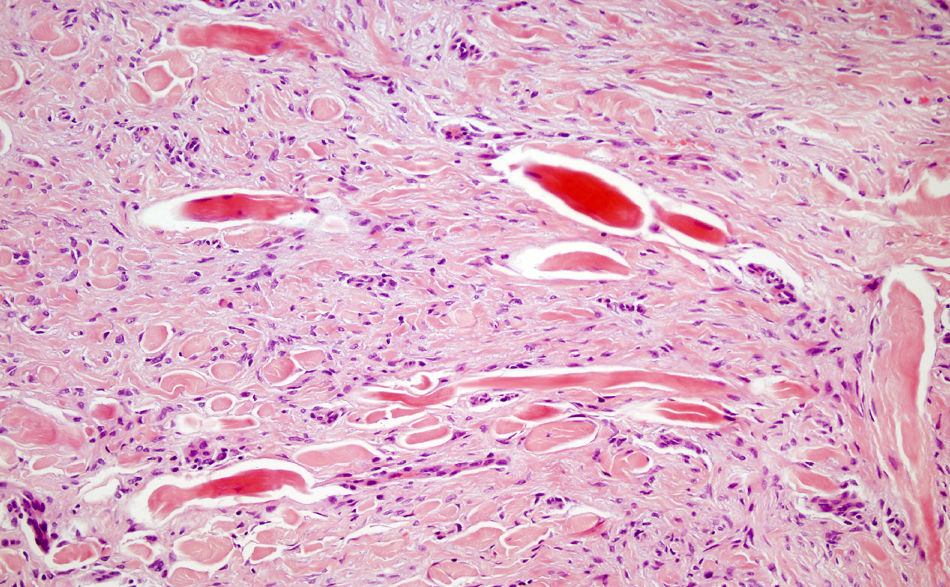
La biopsia de una de las lesiones mostraba hiperplasia epidérmica, bajo la que se identificaba una dermis reticular ocupada por una proliferación de células fusiformes organizadas en fascículos o remolinos celulares, entremezclados con haces de colágeno engrosado (fig. 2). Con las técnicas de inmunohistoquímica se observó una intensa expresión difusa para vimentina y factor XIIIa. La actina de músculo liso, actina muscular específica y el CD34 fueron rigurosamente negativos. Tras el diagnóstico histopatológico de dermatofibroma decidimos abstención terapéutica y seguimiento.
Hemos encontrado 13 casos descritos en la literatura de DF múltiples agrupados, la mayoría recogidos en la revisión de Gershtenson4. Las lesiones individuales no se diferencian clínica, histológica ni fenotípicamente del DF aislado. No describen desencadenantes locales y, a diferencia de los DF múltiples, no se ha encontrado relación con estado de inmunosupresión u otras comorbilidades. Sólo en una ocasión las lesiones se presentaron sobre una trombosis venosa superficial en un paciente que había sido trasplantado de riñón hacía un mes. En la mayoría de los artículos publicados las lesiones, como en nuestra paciente, han aparecido en miembros inferiores. A diferencia de nuestro caso y de otro congénito5, el resto se ha iniciado entre la primera y tercera décadas de la vida. En todos los pacientes se ha objetivado una evolución benigna, no habiendo sido descrito por el momento ningún caso de malignización ni de metástasis secundaria. Así, Berbis refiere un caso seguido durante 20 años sin complicaciones6, lo que consideramos importante a tener en cuenta de cara al manejo y seguimiento de dicha patología.
En el caso de nuestra paciente la clínica, la distribución de las lesiones y el estudio histopatológico se superponen a los descritos previamente, aunque resalta la edad temprana en la que comenzó el cuadro.
Destacamos, por tanto, una forma de presentación atípica, en forma de lesiones múltiples agrupadas, de una patología muy común como es el DF, así como su evolución benigna en todos los casos descritos hasta el momento.


