



INTRODUCCIÓN
La lipoidoproteinosis es una enfermedad caracterizada por el depósito de un material hialino que afecta fundamentalmente a la piel y a las membranas mucosas, aunque también se ha descrito la afectación visceral1 . Recibe también los nombres de hyalinosis cutis et mucosae o enfermedad de Urbach y Wiethe2 . Es una entidad poco frecuente, de distribución mundial, con unos 300 casos publicados en la literatura médica3 . No presenta una predisposición racial, aunque se ha descrito un gran número de casos en África, en posible relación con un inmigrante alemán4 . Afecta por igual a ambos sexos con un patrón de herencia autosómico recesivo4 y se observa consanguinidad de los progenitores en el 20 % de los casos5 . La enfermedad sigue en general un curso progresivo con una esperanza de vida normal4 .
Se presenta un caso de lipoidoproteinosis en una mujer de 23 años.
DESCRIPCIÓN DEL CASO
Una mujer de 23 años, hija de padres no consanguíneos asintomáticos, con 4 hermanos asintomáticos refería haber presentado en los primeros meses de vida un llanto ronco y débil, y una voz ronca desde la infancia. En la piel presentaba lesiones queratósicas iniciadas en la espalda con afectación progresiva de otras localizaciones, a las que se asociaban dificultades para la deglución y la aparición periódica de aftas dolorosas en el dorso lingual.
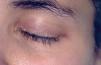
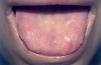
A la exploración física se observaban placas verrugosas en la región lumbar, los codos y las axilas (fig. 1). En ingles, dorso de manos y caras laterales de los dedos presentaba pápulas queratósicas del color de piel normal con superficie lisa, así como hiperqueratosis difusa en las plantas. En el borde libre de los párpados superiores se observaban pápulas blanquecinas con una distribución arrosariada (fig. 2). En las comisuras labiales se observaban pliegues queratósicos. La cavidad oral mostraba queratinización del dorso y raíz linguales con aspecto leñoso y pérdida de las papilas (fig. 3) y las mucosas yugales presentaban un aspecto empedrado; además se observaba estrechamiento del istmo de las fauces y retracción de la úvula. No se observaba ninguna alteración en las uñas, el pelo ni en la zona genital. En la exploración laríngea se observaba una hipertrofia marcada de la epiglotis y de la comisura posterior, con engrosamiento y presencia de nódulos en las cuerdas vocales, y una mucosa faríngea y laríngea edematosa y congestiva. En la revisión oftalmológica se observó triquiasis, con el resto de la exploración normal. Las exploraciones ginecológica y neurológica no presentaron alteraciones.

Fig. 1.—Placas papilomatosas queratósicas excrecentes localizadas en codo.
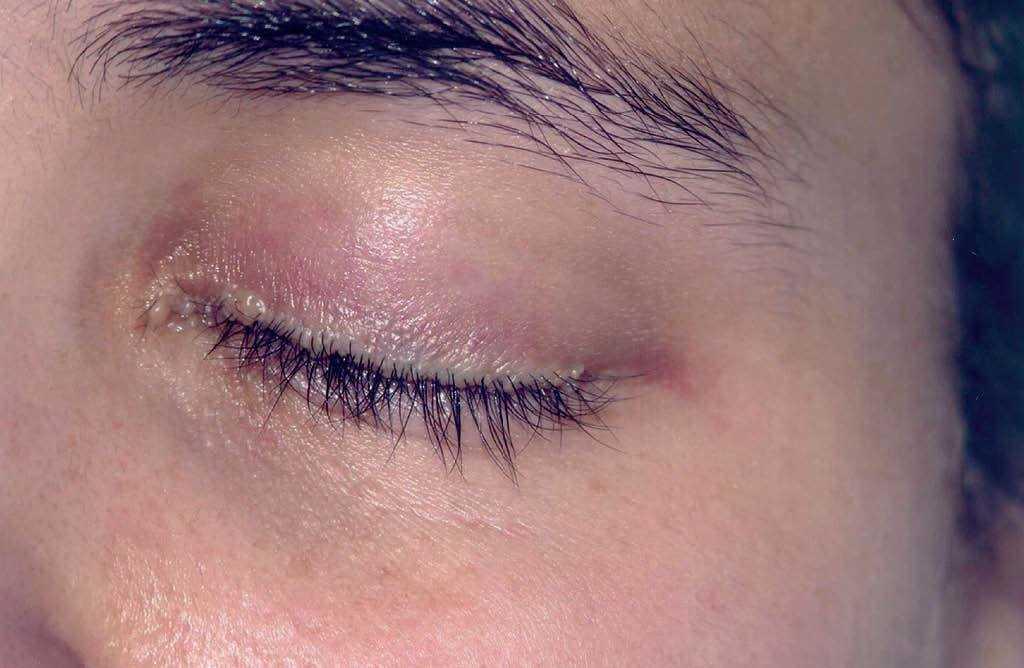
Fig. 2.—Pápulas blanquecinas de distribución lineal con morfología arrosariada en el borde libre del párpado superior.
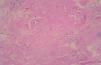
En las pruebas complementarias, el sistemático de sangre, la bioquímica, el sistemático de orina, la coagulación, el proteinograma y las porfirinas en orina mostraron valores normales. La resonancia magnética cerebral presentaba calcificaciones de ambos lóbulos temporales en su porción más anterior, por delante de los cuernos temporales. Se realizó una biopsia cutánea que mostró una epidermis de aspecto verrugoso con hiperqueratosis y una dermis papilar expandida con acumulación de material hialino acelular eosinófilo (fig. 4) rodeando los capilares, los folículos, los ductos ecrinos en la dermis profunda y los músculos erecto-res del pelo. No se observaban infiltrados inflamatorios. El material hialino era PAS positivo resistente a diastasa. El tratamiento tópico con ureal al 40 % y dimetil sulfóxido en solución acuosa al 90 %, ha mostrado buena tolerancia y respuesta discreta.
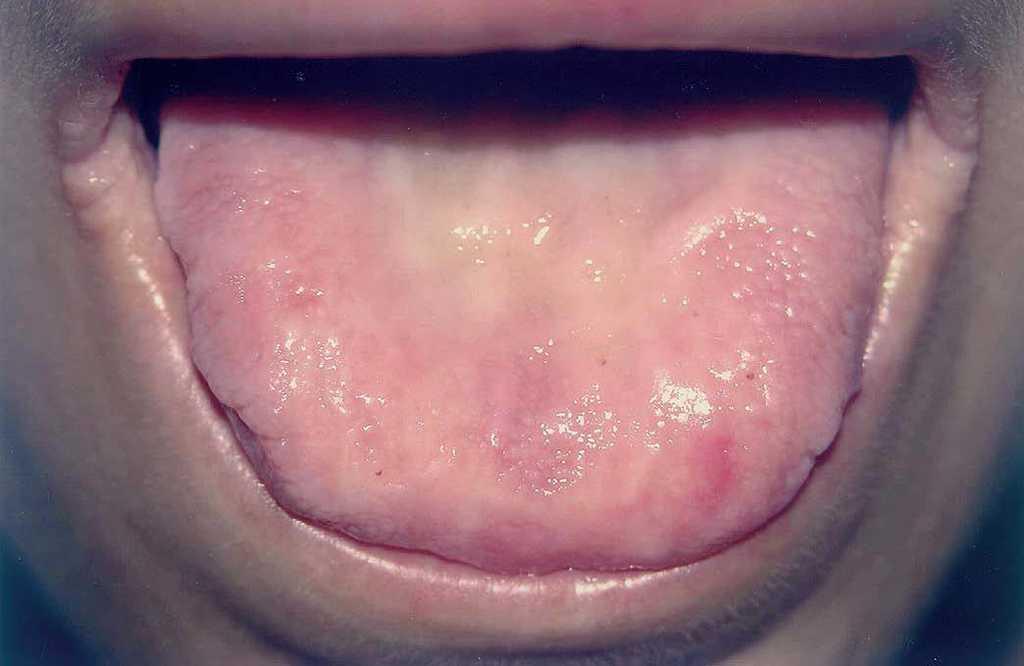
Fig. 3.—Dorso lingual con aspecto leñoso, presencia de hiperqueratosis, pérdida de papilas y dificultad a la protrusión. Fisuras radiales en comisuras labiales.
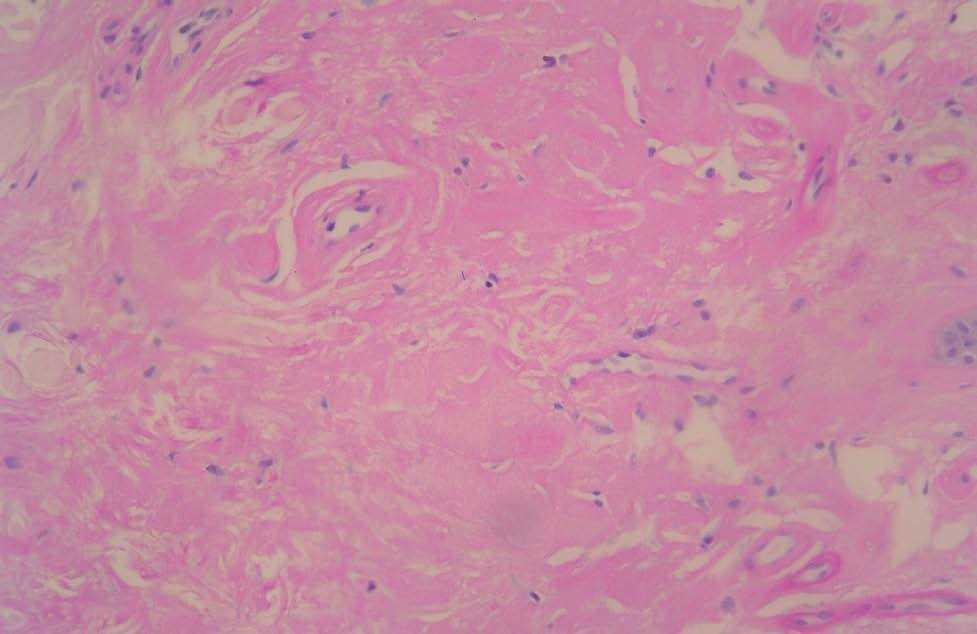
Fig. 4.—Material hialino eosinófilo acelular en la dermis y rodeando los vasos sanguíneos. (Hematoxilina-eosina, ×80.)
DISCUSIÓN
La lipoidoproteinosis fue descrita inicialmente por Siebenmann en 19086 , aunque fueron el dermatólogo Urbach y el otorrinolaringólogo Wiethe quienes acuñaron el término de “lipoidosis cutis et mucosae”2 . Las manifestaciones de esta enfermedad son debidas al depósito de un material hialino amorfo PAS positivo resistente a diastasa7 , que se localiza fundamentalmente en la piel y mucosas. Se ha descrito la presencia del material en órganos internos8 , aunque suele tratarse de un hallazgo de la autopsia1 , porque el depósito es generalmente asintomático4 .
La edad al diagnóstico de la enfermedad es muy variable, desde los 6 meses hasta los 60 años, aunque las manifestaciones clínicas comienzan en la infancia, y afectan por igual a ambos sexos9 . El primer signo de la enfermedad suele ser un llanto ronco y débil desde el nacimiento10 . La ronquera se mantiene con el paso del tiempo, y puede empeorar en climas húmedos10 . La afectación de las cuerdas vocales se produce en el 75 % de los casos10 . Se observan también la mucosa oral queratinizada, la fijación del frenillo lingual que provoca una disminución de la movilidad y la presencia de ulceraciones orales11 . La sintomatología cutánea se inicia generalmente en los primeros 2 años de vida4 , en dos estadios que se superponen. El estadio vesiculoso-erosivo cursa con la presencia de vesículas, erosiones y costras hemorrágicas localizadas en miembros superiores, tronco y cara; las lesiones aparecen ante un mínimo trauma
o fricción y se resuelven espontáneamente dejando una cicatriz deprimida12 . El estadio hiperqueratósico se manifiesta con lesiones engrosadas y amarillentas de aspecto céreo, nódulos y placas en la cara y las flexuras12 . Es característica de la enfermedad, y en el 50 % de los casos se presentan pápulas blanquecinas en el borde libre de los párpados, que reciben el nombre de blefarosis moniliforme5 . También puede asociar alopecia no cicatrizal en la región occipital y fisuras radiales en las comisuras labiales5 . En cuanto a la sintomatología neurológica, pueden presentarse convulsiones, defectos de memoria y ataques de rabia11 . Destaca la presencia de calcificaciones intracraneales en el lóbulo temporal en forma de coma invertida, a la altura de la silla del esfenoides13 ; se trata de un hallazgo patognomónico que generalmente aparece en mayores de 10 años4 .
El diagnóstico de la enfermedad se basa en los hallazgos clínicos, confirmados histológicamente. En la biopsia cutánea destaca la presencia de un material hialino amorfo PAS positivo en la dermis papilar, la membrana basal y alrededor de los capilares14 . Este depósito se observa en microscopia electrónica en forma de anillos concéntricos alrededor de los vasos sanguíneos, con reduplicación de la lámina densa y vacuolización del citoplasma de los fibroblastos14 .
El tratamiento de la lipoidoproteinosis resulta poco satisfactorio. Se emplean corticoides orales para el estadio vesiculoso-erosivo12 . En el estadio hiperqueratósico se han empleado el dimetilsulfóxido por vía oral, la penicilamina y la dermoabrasión15-18 . La respuesta con los distintos tratamientos es variable y el número de pacientes tratados es demasiado pequeño para obtener conclusiones satisfactorias.
Recientemente se ha identificado el defecto genético de la lipoidoproteinosis en el gen que codifica la proteína de matriz extracelular ECM1, localizado en 1q21 19 . El ECM1 es una glucoproteína de 85 kDa de función desconocida, aunque se cree que posiblemente actúa como un “pegamento biológico” en la dermis, manteniendo la homeostasis 14 . El gen de la ECM1, de 10 exones, presenta dos isoformas: ECM1a y ECM1b 19 . Ambas se expresan en la piel y el tracto respiratorio superior 19 . La mayor parte de las mutaciones se han identificado en los exones 6 y 7 y, por tanto, en la isoforma ECM1a, ya que la isoforma ECM1b carece de exón 7 20 . Se ha observado que las mutaciones fuera del exón 7 se asocian a formas clínicas más graves de la enfermedad 20 y no se ha encontrado hasta la fecha una correlación entre genotipo y fenotipo en relación a los síntomas neurológicos 20 .
Correspondencia:
Ana Miguélez. Sección de Dermatología. Hospital Universitario Son Dureta. Avda. Andrea Doria, 55. 07014 Palma de Mallorca. España. anamig@telefonica.net
Recibido el 21 de septiembre de 2004. Aceptado el 17 de febrero de 2005.


