






La leucemia/linfoma de células T del adulto (LLCTA) es una neoplasia agresiva de los linfocitos T asociada a la infección del virus humano T linfotrófico tipo1 (HTLV-1). El HTLV-1 es un problema de salud pública ya que es endémico en etnias nativas de América del Sur, y su infección conlleva diversas enfermedades crónicas, tal como la LLCTA. Nuestro objetivo fue revisar la literatura más reciente sobre LLCTA para su consideración como diagnóstico diferencial ante hallazgos clínicos compatibles. El pronóstico de esta enfermedad es aún malo en las variantes agresivas e indolentes, con sobrevida desde meses a pocos años. El tratamiento, que actualmente se basa en quimioterapia, antirretrovirales y trasplante alogénico de médula ósea, ha mejorado dichos índices de sobrevida, pero aún con resultados limitados.
Adult T-cell Leukemia/Lymphoma (ATLL) is an aggressive neoplasm of T lymphocytes associated with Human T-lymphotropic virus type1 (HTLV-1) infection. HTLV-1 is a public health problem because it is endemic in native groups in Latin America, and its infection leads to several chronic diseases as ATLL. We aimed to review current literature of ATLL in order to consider it as a differential diagnosis in front of patients with compatible symptoms. Prognosis is still poor in aggressive and indolent variants, with survival rates from months to few years. Treatment based on chemotherapy, antiretroviral, and allogenic stem cell transplantation are currently improving survival rates, but with limited results.
La leucemia/linfoma de células T del adulto (LLCTA) es una neoplasia linfoide agresiva de linfocitos T maduros con fenotipo CD4 y CD25 secundaria a infección por virus linfotrópico humano de células T tipo1 (HTLV-1)1.
Aproximadamente, 20 millones de personas se infectan de HTLV-1, en todo el mundo, y la incidencia de la LLCTA en portadores de HTLV-1 alcanza niveles de 8,7 por cada 10.000 portadores en regiones endémicas de Japón2. Por otro lado, el 1% de la población adulta sana de Latinoamérica son portadores asintomáticos3; no obstante, el virus es endémico en algunos grupos amerindios, en quienes la seroprevalencia llega al 10%, igual que ocurre en nativos oriundos de las montañas del sur de Perú4.
En Latinoamérica, donde el virus es más prevalente en grupos nativos con las rentas más bajas, el diagnóstico suele llegar tarde y el pronóstico no es muy alentador. En Japón, el panorama es otro, y la endemicidad del HTLV-1 ha llevado a mejorar tanto la investigación clínica como el pronóstico de pacientes con LLCTA. De ahí que los dermatólogos deban reconocer la verdadera dimensión de esta compleja enfermedad para establecer un diagnóstico precoz, instaurar pronto un tratamiento y ampliar la investigación a pacientes locales en ensayos más extensos.
En esta revisión, hemos intentado exponer la literatura médica existente en la actualidad, incluida la patogénesis, el pronóstico y el tratamiento de la LLCTA.
EpidemiologíaUchiyama et al. describieron, por primera vez, en Japón, la LLCTA en 1977, pero no sería hasta finales de la década de 1980 cuando los estudios seroepidemiológicos establecieron la relación entre esta y el HTLV-15. La proporción de LLCTA entre todos los casos registrados de linfoma entre 2003 y 2008 fue del 8,3% en Japón y del 0,2% en los EE.UU.
Los índices de incidencia estandarizados por la edad de la LLCTA, tanto en Japón como en EE.UU., en 2008 fueron del 0,3 y del 0,02, respectivamente6. Parece que la incidencia de la LLCTA es mucho mayor en zonas no endémicas7, principalmente, debido a la emigración8.
En Japón, la LLCTA sobreviene en torno a los 60 años de edad, con un riesgo acumulativo de desarrollar la enfermedad en torno al 2-5% entre portadores del HTLV-1. Fuera de Japón, la media de edad se sitúa en torno a los 40 años y el riesgo acumulativo está cercano al 4%9.
La mayoría de portadores del HTLV-1 de todo el mundo viven en zonas endémicas, incluido el sudoeste de Japón, algunas islas del Caribe y Sudamérica10. En Japón, la seroprevalencia del HTLV-1 entre 2006 y 2007 se calculó en torno al 0,66 y al 1,02% en varones y mujeres, respectivamente11. En EE.UU. y Europa, se sitúa por debajo del 1%, y se da, principalmente, entre inmigrantes de países endémicos12. En Sudamérica, el virus es predominante en poblaciones étnicas: inmigrantes de otros focos endémicos (como, por ejemplo, África y Japón) y descendientes de nativos amerindios sudamericanos13. En Perú, la prevalencia entre las poblaciones de regiones del sur de los Andes llega al 10%. No obstante, se ha descrito una prevalencia del 17,6 y 21% en 966 y 467 trabajadoras sexuales de Lima y Callao (el puerto más importante), respectivamente14.
Las vías más importantes de transmisión del HTLV-1 se encontraron de madre a hijo, predominantemente, a través de la lactancia materna, las relaciones sexuales y el contacto sanguíneo10,12. La LLCTA está asociada a la lactancia materna (debido a un modelo multipaso de la carcinogénesis)15. Además, sujetos con haplotipos HLA A26, B4002, B4006 y B4801 parecen estar genéticamente más predispuestos a desarrollar LLCTA5.
PatogénesisEl HTLV-1 es un retrovirus que codifica 3 proteínas estructurales: Gag, Pol y Env. La proteína Pol codifica las funciones de la transcriptasa inversa, la proteasa y la integrasa. La proteína Gag proporciona al virión proteínas básicas y la Env se utiliza a efectos de infectividad viral. Además, el genoma del HTLV-1 tiene una región pX que codifica proteínas reguladoras como Tax y Rex, entre otras12. La transcriptasa inversa genera ADN proviral procedente del ARN viral genómico tras la transmisión. La integrasa viral logra integrar el ADN en el genoma anfitrión. Después, el HTLV-1 genera pocas o ninguna partícula similar a virus; sin embargo, la infección se propaga por íntimo contacto célula a célula16.
La carcinogénesis del HTLV-1 es un proceso bifásico (fig. 1). Primero, la proteína reguladora Tax juega un papel crucial en la supervivencia, proliferación y transformación de las células infectadas con HTLV-1 in vivo. Solo la expresión de la Tax puede bastar para la inmortalización de las células T humanas, principalmente, células T reguladoras, que provocan la formación de tumores in vivo17. El papel que juega la proteína reguladora Tax en la supervivencia, proliferación y oncogénesis se debe a varias acciones como son: (i) la activación de NF-kB y Akt (2 importantes vías de prosupervivencia celular), (ii) la inactivación de la proteína p53, (iii) el daño sufrido por el ADN y ocasionado tanto por lesiones directas como por la atenuación de la sensibilidad al daño y de las proteínas reparadoras, (iv) la inducción de especies de oxígeno reactivo, y (v) los cambios en los microARN con potenciales oncogénicos18. Por todo esto, la proteína Tax es crucial tanto para la propagación de células infectadas con HTLV-1 como para la transformación original en las fases iniciales, si bien es inmunogénica y solo se encuentra en el 40% de las células LLCTA18.

Patogénesis de la LLCTA. Infección de células T reguladoras por contacto célula a célula. La carcinogénesis es un proceso bifásico. Primero, la proteína reguladora Tax está implicada en la transformación maligna inicial, pero solo está presente en el 40% de las células LLCTA. De ahí que el factor HBZ juegue un papel primordial en la proliferación, mantenimiento y evasión inmunológica.
HBZ: factor de cremallera de leucina de carácter básico del HTLV-1; HTLV-1: infección del virus humano T linfotrófico tipo1; LLCTA: leucemia/linfoma de células T del adulto; TAX: producto del gen Tax.
Tras desarrollar la inmunidad, las células T citotóxicas CD8+ son capaces de eliminar las células que expresan la Tax y de contener, por tanto, la infección. Después, las células infectadas con HTLV-1 pueden proliferar por la continua expresión del factor de cremallera de leucina de carácter básico (HBZ) del HTLV-1, expresado ubicuamente en todas las células LLCTA después de la Tax, con niveles más altos con el paso del tiempo11,19. El HBZ juega un papel importante tanto en la replicación viral como en la proliferación, el mantenimiento y la evasión inmunológica de las células LLCTA. De este modo, el HBZ transcribe el mARN viral uniformemente expresado en las células LLCTA, atenúa las respuestas inmunológicas del anfitrión para evitar la destrucción de las células LLCTA y modifica un microambiente favorable para el HTLV-111,20.
Por lo tanto, el HBZ antagoniza y complementa muchas de las actividades de la Tax. La Tax entra pronto en escena, mientras que el HBZ lo hace más adelante durante la infección del virus. La primera se utiliza para iniciar la transformación (efecto ocogénico) y el HBZ para mantener el fenotipo transformado de células LLCTA21 (fig. 1).
Aunque el HTLV-1 infecta células T, células B, fibroblastos, células dendríticas y macrófagos, se considera que solo las células T reguladoras que expresan el CD25 y el factor de transcripción de la familia de la caja de la cabeza de tenedor P3 (FoxP3) son el tipo de células que se transforman en LLCTA5. La expresión del FoxP3 es importante porque tiene efectos beneficiosos y perjudiciales por igual: suprime el crecimiento de clones LLCTA autógenos pero, también, la respuesta de los linfocitos T citotóxicos del anfitrión, que suele limitar la replicación del HTLV-1 y minimizar el riesgo de LLCTA11,22.
ClasificaciónLa LLCTA fue subclasificada en 4 variantes según la Clasificación de Tumores de Tejidos Hematopoyéticos y Linfoides de la Organización Mundial de la Salud (OMS) en 2008, atendiendo a la clasificación de Shimoyama, en base al compromiso de los órganos, al lactato deshidrogenasa (LDH) y a los valores de calcio. Las variantes son: aguda (60%), linfomatosa (20%), crónica (15%) y quiescente (5%)23,24.
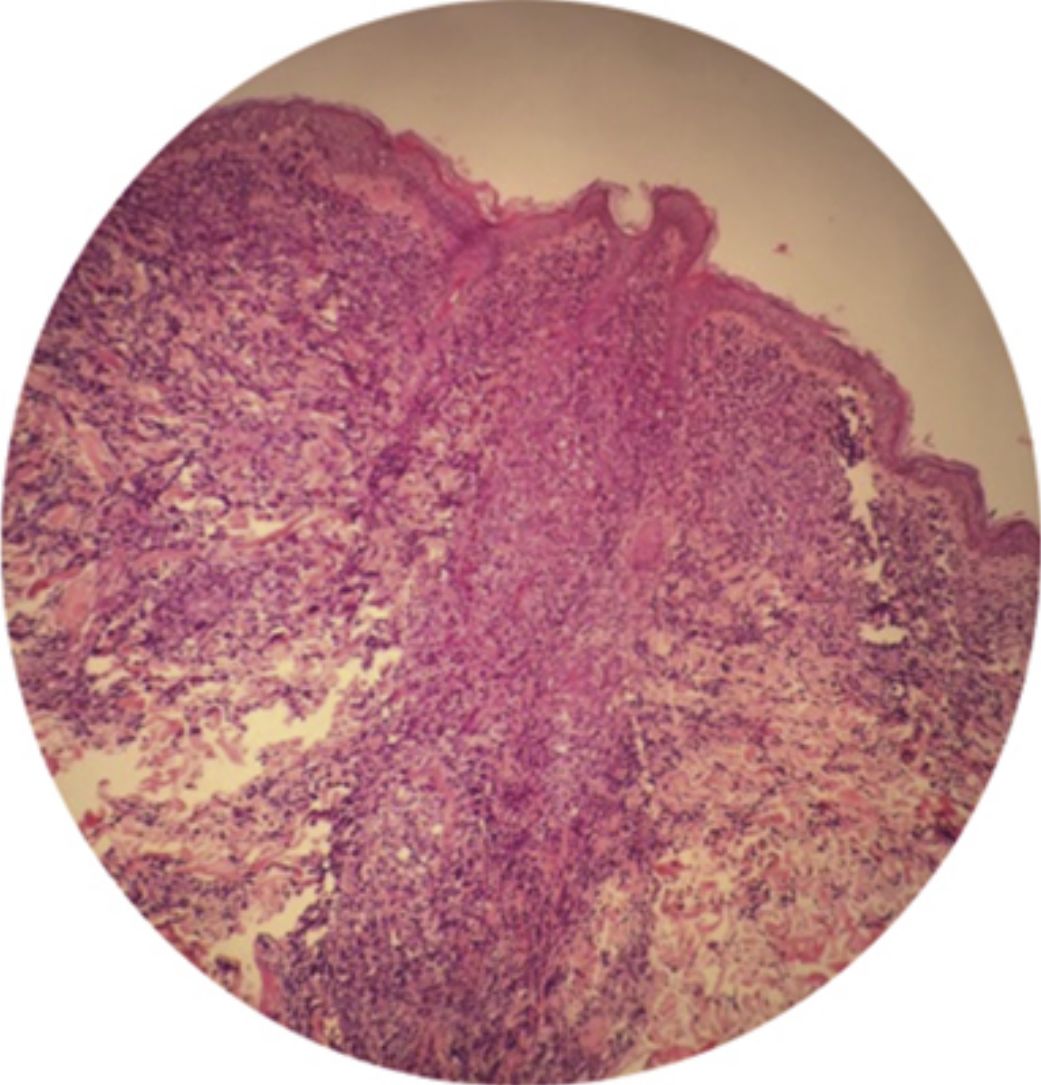
La variante aguda es la más agresiva y se caracteriza por presentar una marcada leucocitosis con linfocitos atípicos, lesiones cutáneas (erupción eritematosa, nódulos o pápulas), síntomas constitucionales, linfadenopatía masiva sin afectación del mediastino, y hepatosplenomegalia25. Suele haber hipercalcemia y niveles altos de LDH. Puede haber complicaciones respiratorias secundarias a infiltración tumoral o infecciones oportunistas y sepsis5 (fig. 2).
 Mujer de 64 años de edad con múltiples pápulas distribuidas por tronco y extremidades proximales de 3 meses de evolución. (b) Las pápulas son asintomáticas, eritematosas y con una buena consistencia. (c) Infiltración de la dermis por células linfoides atípicas con epidermotropismo. Tinción con hematoxilina y eosina, ×20. (d) Inmunohistoquímica CD3+, ×20. La paciente falleció 2 semanas después del ingreso a causa de un shock séptico.")
LLCTA aguda. (a) Mujer de 64 años de edad con múltiples pápulas distribuidas por tronco y extremidades proximales de 3 meses de evolución. (b) Las pápulas son asintomáticas, eritematosas y con una buena consistencia. (c) Infiltración de la dermis por células linfoides atípicas con epidermotropismo. Tinción con hematoxilina y eosina, ×20. (d) Inmunohistoquímica CD3+, ×20. La paciente falleció 2 semanas después del ingreso a causa de un shock séptico.
La variante linfomatosa también es agresiva, pero su característica más prominente es una marcada linfadenopatía sin leucemia. Tanto el compromiso cutáneo como la hipercalcemia son menos frecuentes25,26.
La variante crónica suele presentarse con erupciones cutáneas exfoliativas, leucocitosis menos prominente con linfocitosis absoluta, linfadenopatía de carácter leve e hipercalcemia25 (figs. 3 y 4).
 Varón de 91 años de edad con múltiples placas pruríticas y eritematosas en tronco de un año de evolución, asociadas a un cuadro de xerosis severa. (b) Epidermotropismo y absceso de Pautriers. Tinción con hematoxilina y eosina, ×40. (c) Inmunohistoquímica CD3+, ×40. El paciente falleció 4 meses después del ingreso. Estuvo en tratamiento paliativo y sintomático.")
LLCTA crónica. (a) Varón de 91 años de edad con múltiples placas pruríticas y eritematosas en tronco de un año de evolución, asociadas a un cuadro de xerosis severa. (b) Epidermotropismo y absceso de Pautriers. Tinción con hematoxilina y eosina, ×40. (c) Inmunohistoquímica CD3+, ×40. El paciente falleció 4 meses después del ingreso. Estuvo en tratamiento paliativo y sintomático.
 Varón de 66 años de edad con eritroderma y xerosis severa de 3 años de evolución. (b) Epidermotropismo y microabscesos de Pautriers. Tinción con hematoxilina y eosina, ×40. (c) Inmunohistoquímica CD3+, ×40. El paciente falleció 8 meses después de empezar un tratamiento con quimioterapia con gemcitabina (1.000mg/m2 cada día) y oxaliplatino (100mg/m2 cada día) cada 3 semanas.")
LLCTA crónica. (a) Varón de 66 años de edad con eritroderma y xerosis severa de 3 años de evolución. (b) Epidermotropismo y microabscesos de Pautriers. Tinción con hematoxilina y eosina, ×40. (c) Inmunohistoquímica CD3+, ×40. El paciente falleció 8 meses después de empezar un tratamiento con quimioterapia con gemcitabina (1.000mg/m2 cada día) y oxaliplatino (100mg/m2 cada día) cada 3 semanas.
La variante quiescente suele ser asintomática. Los pacientes suelen presentar un recuento normal de glóbulos blancos con menos del 5% de células neoplásicas circulantes y ausencia de hipercalcemia u organomegalia asociadas. Al igual que ocurre con la variante crónica, el tipo quiescente acarrea un riesgo del 25% de pasar a la fase aguda5.
Hallazgos clínicosLas lesiones cutáneas se dan en el 43-72% de todas las variantes clínicas de la LLCTA debido a la invasión directa de células malignas en la piel, resultando en diferentes tipos de erupciones. Se han descrito 6 tipos de erupciones: nodulotumorales (38,7%), en placa (26,9%), multipapulares (19,3%), en parche (6,7%), eritrodérmicas (4,2%) y purpúricas (4,2%). Las erupciones cutáneas no son exclusivas y suelen ser crónicas, indoloras o pruríticas27,28. Las erupciones de carácter general se dan en el 70%, el tronco se ve comprometido en el 10% de los casos, el tronco y las extremidades en el 25%, y, solo, la cabeza, el cuello, o las extremidades en el 5%29 (figs. 2-4).
Se han descrito formas raras de compromiso cutáneo por LLCTA, tales como lesiones de tipo eccematoso dishidrótico en manos y pies, lesiones queloides, lesiones de tipo granulomatoso y un caso con lesiones vesiculares y ampollosas30. Además, otras formas de presentación descritas son lesiones de tipo eccematoso, esclerodérmico e ictiosiforme31.
Cabe destacar que el tipo de erupción cutánea por LLCTA presente se ha identificado como un factor pronóstico independiente. Los parches y las placas se asocian a una mejor supervivencia, mientras que las presentaciones eritrodérmicas y nodulotumorales tienen un peor pronóstico28.
Hallazgos en laboratorio y diagnósticoLa histología revela 3 patrones distintos de infiltración: perivascular, nodular y difuso. El epidermotropismo y los microabscesos de Pautrier son una característica frecuente32 (figs. 2-4). Como estos hallazgos nos recuerdan a otros linfomas de células T cutáneos, el análisis inmunohistoquímico juega un papel esencial a la hora de realizar un diagnóstico diferencial. Las células LLCTA expresan marcadores de células T maduras tales como CD2, CD5, CD25 (fuerte y uniforme), CD29, CD45RO, TCR y HLA-DR, aunque también suelen expresar CD4+ y CD8–33. La expresión del CD25 (receptor de interleucina 2) puede ser característica pero no específica, ya que también puede presentarse en la leucemia prolinfocítica T y en el síndrome de Sézary (SS)33.
El compromiso de sangre periférica puede detectarse en las variantes aguda y crónica, principalmente con leucocitosis y linfocitosis. El frotis de sangre periférica confirma la presencia de blastos a modo de «células en forma de flor», que son linfocitos de tamaño medio-grande con núcleos con forma de pétalos y un patrón de cromatina áspera, CD4+ y CD25+34. Suelen confundirse con sombras de Gumprecht.
La biopsia de la médula espinal revela infiltración sutil o en forma de parche de células linfoides atípicas con contornos nucleares irregulares (fig. 2). No obstante, no es imperativo para el diagnóstico de LLCTA35. Además, el compromiso de la médula espinal >5% de las células LLCTA en el aspirado o biopsia se considera un mal factor pronóstico independiente5. Los nódulos linfáticos comprometidos suelen exhibir compromiso difuso, principalmente, con expansión paracortical36.
El análisis de flujo citométrico de células T en la LLCTA revela que esta es una malignidad de células T reguladoras/células Th2 con expresión de alta frecuencia de CD3/CD4/CD25/CCR4 y FoxP3 en casi la mitad de las células.
El diagnóstico de LLCTA se basa en una combinación de características clínicas, cambios morfológicos e inmunofenotípicos de las células malignas, en la confirmación de infección por HTLV-1, así como en la realización de pruebas en busca de anticuerpos específicos y/o la realización de la prueba de la reacción en cadena de la polimerasa (RCP) en células de sangre periférica35,37. La identificación de, al menos, un 5% de células tumorales en la citología y en el inmunofenotipo en sangre periférica acompañado de la confirmación de infección por HTLV-1 suele bastar para poder llegar al diagnóstico en pacientes con tipos agudos, crónicos o quiescentes de LLCTA. La variante linfomatosa necesita de la realización de una biopsia excisional de algún nódulo linfático comprometido para su posterior análisis histopatológico38. La integración del provirus clonal en laboratorio no suele ser habitual; de ahí que el diagnóstico suela confirmarse.
El diagnóstico diferencial patológico de la LLCTA incluye linfoma T periférico no especificado (LTPNE), linfoma anaplásico de células grandes, linfomas cutáneos de células T tales como micosis fungoide (MF) y SS y linfoma de células T angioinmunoblástico. El LTPNE es el tipo más común de linfoma de células T que existe en Norteamérica y puede ser patológicamente indistinguible de la LLCTA. Tanto la MF como el SS son importantes factores diferenciales a tener en cuenta, ya que se observan similares hallazgos clínicos e infiltración histológica de linfocitos. Si no se obtiene del paciente confirmación de que ha viajado a una de las regiones endémicas, el diagnóstico de LLCTA puede pasarse por alto. Es muy fácil pasar por alto este diagnóstico en regiones de baja prevalencia, si bien una serología positiva por HTLV-1 o una confirmación de integración viral en células neoplásicas serán, siempre, favorables a LLCTA en todos los casos39.
Pronóstico y predictores de resultadosPor lo general, el pronóstico de la LLCTA es peor que el del LTPNE. Las variantes agresivas, aguda y linfomatosa son las que peor pronóstico tienen, con un tiempo medio de supervivencia (TMS) de entre 6 y 10 meses, respectivamente. Entre tanto, las variantes indolentes como, por ejemplo, las variantes crónica y quiescente son las que tienen un TMS más largo, de casi 2 años y más de 2 años, respectivamente25,36. La muerte suele sobrevenir por complicaciones infeccionas, hipercalcemia incontrolada o enfermedad progresiva36.
Se han desarrollado varios sistemas pronósticos para identificar a aquellos grupos que tienen un pronóstico mucho mejor. Se agruparon a 807 pacientes según factores pronósticos tales como el estadio clínico Ann Arbor (I y II vs. III y IV), el estado de rendimiento (ER) (de 0 a 1 vs. de 2 a 4), la edad, la albúmina en suero y el receptor soluble de interleucina 2 (CD25). Los autores calcularon una función de riesgo lineal llamada LLCTA-PI basada en los coeficientes de regresión de Cox del siguiente modo: LLCTA-PI=0,65 (con un estadio=III o IV)+0,35 (con un ER >1)+0,016×edad (años)–0,36×albúmina (g/dl)+0,37×log10 (receptor de interleucina 2[U/ml]). El grupo de riesgo bajo fue aquel con una LLCTA-PI <1,6; el grupo de riesgo intermedio, como 1,6≤LLCTA-PI >2,6; y el grupo de riesgo alto fue aquel con una LLCTA-PI≥2,6. El TMS fue de 3,6, 7,3 y 16,2 meses, en los grupos de riesgo alto, intermedio y bajo, respectivamente40.
El Grupo Japonés de Oncología Clínica (JCOG) y el Grupo para el Estudio de los Linfomas (LSG) desarrollaron 2 importantes factores pronósticos: ER e hipercalcemia. El grupo de riesgo moderado (valores de calcio corregido <2,75 mmol/l y un ER de 0 o 1) tiene un TMS y una supervivencia global (SG) a los 5 años de 14 meses y del 18%, respectivamente. El grupo de riesgo alto (valores de calcio corregido ≥2,75mmol/l y/o un ER de 2-4), un TMS y una SG a los 5 años de 8 meses y del 4%, respectivamente41.
Como se asocian diferentes variantes con diferentes factores pronósticos, un estudio de 95 pacientes peruanos con LLCTA extrajo varias puntuaciones útiles para estratificar el riesgo de pacientes con variantes agresivas de LLCTA42. Los niveles bajos de albúmina y la presencia de síntomas B fueron factores independientes de una peor supervivencia en la LLCTA linfomatosa, siendo el nivel de microglobulina beta 2 un factor independiente de una peor supervivencia en la variante aguda de la LLCTA42.
Cabe destacar que algunos factores pronósticos también pueden dictar las decisiones sobre el tratamiento a instaurar. Aquí encontramos factores clínicos tales como el ER, el LDH, la edad, el estadio, el número de lesiones y la hipercalcemia, y factores moleculares tales como la expresión del antígeno Ki-67, el receptor soluble de interleucina 2, la alteración de la proteína p53 y la sobreexpresión del factor-4 regulador del interferón (IRF-4)11.
TratamientoEn términos generales, los estudios confirman excelentes indicadores de supervivencia con tratamiento con zidovudina/interferónα (AZT/IFN) en pacientes con LLCTA indolente, y quimioterapia multifármaco y/o trasplante alogénico de células madre hematopoyéticas (TACMH) para las formas agresivas en Japón. En países occidentales, el tratamiento con AZT/IFN con o sin quimioterapia es el tratamiento elegido5.
Los recientes regímenes de quimioterapia tales como el régimen LSG15 modificado consisten en la instauración de 6 ciclos de vincristina, ciclofosfamida, doxorrubicina y prednisona (VCAP); doxorrubicina, ranimustina y prednisona (AMP) y vindesina, etopósido, carboplatino y prednisona (VECP). Este régimen generó un índice de supervivencia a los 3 años del 24%, un índice de respuesta completa del 40% y un TMS de 13 meses en 118 pacientes no tratados previamente con la variante agresiva de LLCTA43.
Por otro lado, el tratamiento con AZT/IFN puede ser prometedor, sobre todo para los tipos con manifestaciones leucémicas11. Un reciente metaanálisis reveló un índice de SG a los 5 años del 46% para 75 pacientes que recibieron tratamiento antiviral de primera línea, del 20% para 77 pacientes que recibieron quimioterapia como tratamiento de primera línea y del 12% para 55 pacientes que recibieron quimioterapia como tratamiento de primera línea seguido de tratamiento antiviral44. Cabe destacar que la dosis de INF y de terapia antiviral todavía no se ha estandarizado. Un abordaje es instaurar INFα 2b en dosis de 5MU, por vía subcutánea, durante 8-12 semanas en combinación con 300mg de AZT 3 veces/día. A la finalización del tratamiento con IFNα, el tratamiento con 300mg de AZT 3 veces/día se continúa indefinidamente45.
El TACMH resulta prometedor para el tratamiento de la variante agresiva de la LLCTA, y refleja, posiblemente, un efecto injerto contra LLCTA. En un estudio comparativo de 56 pacientes, aquellos que recibieron quimioterapia tras un TACMH o, solo, quimioterapia, tuvieron un índice de SG a los 3 años del 44,9 y 27,7%, respectivamente46,47. Algunos autores proponen que el TACMH debe considerarse pronto, concretamente 100 días antes del diagnóstico de LLCTA. En 72 pacientes con LLCTA, aquellos en quienes el TACMH se llevó a cabo más precozmente, tuvieron un índice de SG a los 4 años del 49 y 31%, respectivamente48.
El Congreso Internacional de Consenso de la Sociedad Americana de Oncología Clínica publicó las actuales directrices para el manejo de la LLCTA en 200935 (fig. 5). Primero, recomiendan poner las variantes quiescente y crónica favorable de LLCTA en observación, siempre y cuando esta sea asintomática. En las variantes sintomáticas (como, por ejemplo, lesiones cutáneas e infecciones oportunistas) se recomienda instaurar un régimen de AZT/IFN o mantener la LLCTA en observación. En las variantes crónica desfavorable y aguda con pobres factores pronósticos se recomienda instaurar un régimen de quimioterapia con VCAP-AMP-VECP o AZT/IFN. En las variantes agudas con buenos factores pronósticos se recomienda instaurar un régimen de quimioterapia seguido de un TACMH convencional o de intensidad reducida.
.")
Estrategia para el tratamiento de la LLCTA, propuesta por la Sociedad Americana de Oncología Clínica en 200935.
allo-HSCT: trasplante alogénico de células madre hematopoyéticas; AMP: doxorrubicina, ranimustina y prednisona; ATLL/LLCTA: leucemia/linfoma de células T del adulto; AZT/IFN: zidovudina/interferónα; CT: ensayos clínicos; Chemo: quimioterapia; VCAP: vincristina, ciclofosfamida, doxorrubicina y prednisona; VECP: vindesina, etopósido, carboplatino y prednisona.
* La enfermedad favorable y desfavorable se basa en factores pronóstico que incluyen factores clínicos, tales como el estado de rendimiento, el LDH, la edad, el estadio, el número de lesiones implicadas y la hipercalcemia; y factores moleculares, tales como la expresión del antígeno Ki-67, el receptor soluble de interleucina 2, la alteración de la proteína p53 y la sobreexpresión de IRF-4.
** Las opciones con ensayos clínicos incluyen probar el efecto del TACMH, del tratamiento combinado con trióxido de arsénico, IFN, bortezomib, VCAP-AMP-VECP, o el tratamiento antiangiogénico, el IFNα pegilado y AZT y los anticuerpos monoclonales (mogamulizumab).
En pacientes con una mala respuesta al tratamiento inicial con quimioterapia o AZT/IFN se recomienda llevar a cabo un TACMH convencional o de intensidad reducida35.
En la variante linfomatosa se recomienda la instauración de un régimen de quimio con VCAP-AMP-VECP. Si los perfiles pronósticos son favorables y hay una buena respuesta al tratamiento inicial, los pacientes deberían continuar el tratamiento con quimioterapia. No obstante, si los perfiles pronósticos no lo son, o hay una mala respuesta al tratamiento inicial, entonces debe pensarse en llevar a cabo un TACMH convencional o de intensidad reducida35.
El CCR4 está expresado en las células neoplásicas de la mayoría de pacientes con LLCTA, y esta expresión se ha asociado a manifestaciones cutáneas y a un mal pronóstico11. El anticuerpo afucosilado humanizado anti-CCR4 mogamulizumab (Moga) se ha aprobado recientemente en Japón para el tratamiento de la LLCTA refractaria o recidivante. Moga es altamente citotóxico para las células LLCTA a través de la citotoxicidad celular dependiente del anticuerpo y depleta las células T reguladoras durante, al menos, unos cuantos meses49. De ahí que preocupen las influencias negativas que puede tener el pretrasplante del Moga, ya que puede aumentar el riesgo de desarrollar enfermedad injerto contra huésped tras el TACMH50. Además, se han descrito reacciones adversas tales como el síndrome de Stevens-Johnson51. Moga tiene un índice de respuesta de, aproximadamente, el 50% en la LLCTA refractaria/recidivante52. No obstante, recientes ensayos han confirmado la superioridad de un ciclo combinado de Moga con LSG15 modificado frente a solo quimioterapia, para el manejo de la LLCTA agresiva no tratada. El tratamiento combinado generó una respuesta completa más alta del 52% frente al 33%, respectivamente53.
ConclusiónLa LLCTA es una neoplasia agresiva de células T reguladoras provocada por una infección crónica por HLTV-1, de difícil diagnóstico y mal pronóstico. Como el HTLV-1 es endémico de varios grupos nativos de Japón y Latinoamérica, los dermatólogos deben tener en cuenta el diagnóstico de LLCTA en pacientes con erupciones cutáneas compatibles y epidermotropismo con invasión dérmica linfocítica en la histología. Los hallazgos clínicos y de laboratorio definen los distintos subtipos de LLCTA que pueden darse, lo cual está íntimamente ligado al pronóstico. El MF y el SS son los principales factores diferenciales, ya que presentan similares hallazgos clínicos e histológicos. Aunque las variantes indolentes pueden beneficiarse de regímenes de tratamiento AZT/INF, las variantes agresivas pueden responder tanto a la quimioterapia como al TACMH. El anticuerpo monoclonal mogalizumab está aprobado para casos refractarios o recidivas. Los dermatólogos han de ser conscientes de la dimensión que tiene esta compleja enfermedad, ya que la infección por HTLV-1 sigue siendo un problema de salud pública y se destinan pocos recursos y hay pocas investigaciones en marcha encaminadas a resolver este problema en países latinos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Agradecemos a la Dra. Dina Carayhua de la Unidad de Dermopatología su ayuda durante el análisis y recogida de casos histopatológicos.


