




Adult T-cell Leukemia/Lymphoma (ATLL) is an aggressive neoplasm of T lymphocytes associated with Human T-lymphotropic virus type1 (HTLV-1) infection. HTLV-1 is a public health problem because it is endemic in native groups in Latin America, and its infection leads to several chronic diseases as ATLL. We aimed to review current literature of ATLL in order to consider it as a differential diagnosis in front of patients with compatible symptoms. Prognosis is still poor in aggressive and indolent variants, with survival rates from months to few years. Treatment based on chemotherapy, antiretroviral, and allogenic stem cell transplantation are currently improving survival rates, but with limited results.
La leucemia/linfoma de células T del adulto (LLCTA) es una neoplasia agresiva de los linfocitos T asociada a la infección del virus humano T linfotrófico tipo1 (HTLV-1). El HTLV-1 es un problema de salud pública ya que es endémico en etnias nativas de América del Sur, y su infección conlleva diversas enfermedades crónicas, tal como la LLCTA. Nuestro objetivo fue revisar la literatura más reciente sobre LLCTA para su consideración como diagnóstico diferencial ante hallazgos clínicos compatibles. El pronóstico de esta enfermedad es aún malo en las variantes agresivas e indolentes, con sobrevida desde meses a pocos años. El tratamiento, que actualmente se basa en quimioterapia, antirretrovirales y trasplante alogénico de médula ósea, ha mejorado dichos índices de sobrevida, pero aún con resultados limitados.
Adult T-cell Leukemia/Lymphoma (ATLL) is an aggressive lymphoid neoplasm of mature T lymphocytes with CD4 and CD25 phenotype caused by Human T-lymphotropic virus type I (HTLV-I) infection.1
Approximately 20 million people are infected with HTLV-I in the world, and the incidence of ATLL in HTLV-I carriers reaches levels of 8.7 per 10,000 carriers in endemic regions of Japan.2 On the other side, in Latin America 1% of the healthy adult population are asymptomatic carriers3; nevertheless, the virus is endemic in some Amerindian groups, where the seroprevalence reaches 10% as in natives from the southern mountains of Peru.4
In Latin America, where the virus is most prevalent in native and lowest income groups, diagnosis is often delayed with a dismal prognosis. A different scenario is found in Japan, where HTLV-I endemicity in several regions carried to improving clinical research and prognosis of patients with ATLL. Therefore, dermatologists need to recognize the dimension of this complex disease in order to establish an early diagnosis, to offer prompt treatment to patients, and to expand research including local patients into large trials.
In this review, we aimed to expose the currently available literature regarding the pathogenesis, prognosis, and treatment of ATLL.
EpidemiologyUchiyama et al. in Japan first described ATLL in 1977, but it was later in the 1980s when seroepidemiologic studies established its relation with HTLV-1.5 The proportion of ATLL among all registered lymphoma cases in 2003–2008 was 8.3% in Japan and 0.2% in the United States (US). The age-standardized incidence rates of ATLL in Japan and the US in 2008 were 0.3 and 0.02, respectively.6 It seems that the incidence of ATLL is significantly increasing in non-endemic areas7 mostly due to emigration.8
In Japan, ATLL appears at a median age of 60 yo., with a cumulative risk of developing the disease around 2% to 5% among HTLV-1 carriers. Outside Japan, the median age is around 40yo and the cumulative risk near 4%.9
Most of the HTLV-I carriers in the world live in endemic areas including southwestern Japan, some of the Caribbean islands and South America.10 In Japan, the seroprevalence of HTLV-I between 2006 and 2007 was estimated to be 0.66% and 1.02% in men and women, respectively.11 In the US and Europe, it is less than 1%, primarily seen on immigrants from endemic countries.12 In South America, the virus is predominant in ethnic populations: immigrants from other endemic foci (as Africa and Japan) and in those who descend from native South America Amerindians.13 In Peru, prevalence in population from the South Andean region reaches 10%. However, a prevalence of 17.6% and 21% were reported in 966 and 467 sexual workers from Lima and Callao (the main harbor), respectively.14
The most important routes of HTLV-1 transmission were found to be from mother to child, predominantly through breastfeeding; sexual intercourse; and blood contact.10,12 ATLL has been associated with breastfeeding (due to a multistep carcinogenesis model).15 Additionally, individuals with HLA A26, B4002, B4006, and B4801 appear to be genetically more predisposed to develop ATLL.5
PathogenesisHTLV-1 is a retrovirus and encodes three structural proteins: Gag, Pol, and Env. The Pol encodes reverse transcription, protease, and integrase functions. Gag provides the virion core proteins, and Env is used for viral infectivity. In addition, HTLV-1 genome has a pX region that encodes regulatory proteins as Tax, Rex among others.12 Reverse transcriptase generates proviral DNA from genomic viral RNA after transmission. Viral integrase manages to integrate the DNA into the host genome. Afterward, HTLV-1 produces little to no free viral particles; nevertheless, infection is spread by close cell-to-cell contact.16
HTLV-1 carcinogenesis is a 2-phase process (Fig. 1). First, Tax plays a central role in survival, proliferation, and transformation of HTLV-1-infected cells in vivo. The expression of Tax alone may be sufficient for immortalization of human T-cells, mainly regulatory T-cells, driving in vivo tumor formation.17 The role of Tax on survival, proliferation, and oncogenesis is due to several actions. These are: (i) the activation of NF-kB and Akt (two major cellular prosurvival routes); (ii) inactivation of p53 protein; (iii) DNA-damage by direct lesions and attenuation of damage sensing and repair proteins; (iv) induction of reactive oxygen species; (v) and changes in microRNAs with oncogenic potentials.18 Therefore, Tax is crucial for the expansion of HTLV-1–infected cells and original transformation in early phases,11,19 but it is immunogenic and is only present in near 40% of transformed ATLL cells.18
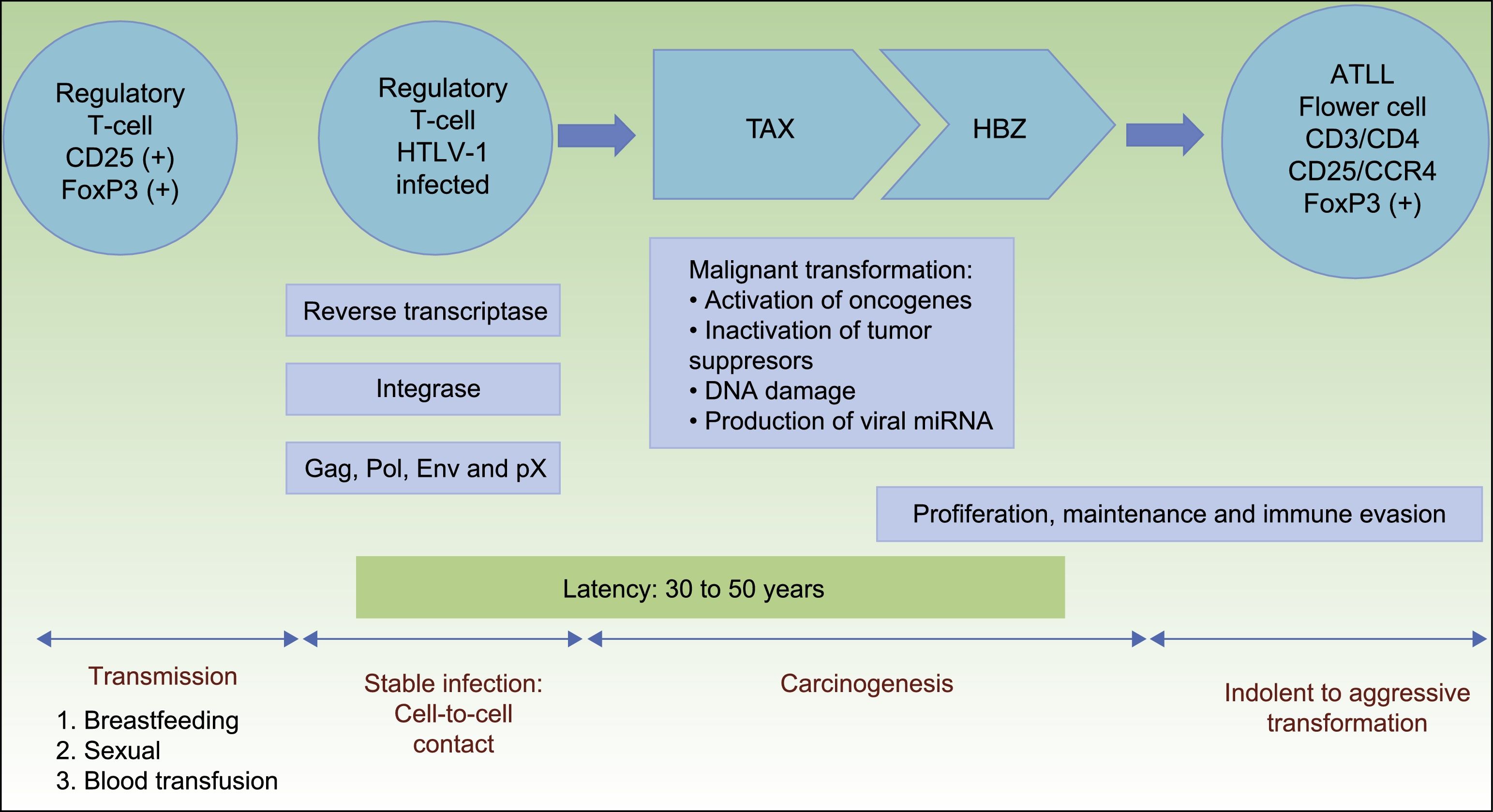
Pathogenesis of ATLL. Infection of regulatory T-cells occurs through cell-to-cell contact. Carcinogenesis is a two-step process. First, the regulatory protein Tax is involved in early malignant transformation, but is only present in 40% of ATLL cells. Thereafter, HBZ plays a role in the proliferation, maintenance and immune evasion.
Abbreviations ATLL: Adult T-cell Leukemia/Lymphoma; TAX: Tax gene product; HBZ: HTLV-1 basic leucine zipper factor.
Once immunity develops, cytotoxic CD8+ T-cell can eliminate the Tax-expressing cells, thus containing the infection. Thereafter, HTLV-1–infected cells can proliferate with the continued expression of the HTLV-1 basic leucine zipper factor (HBZ), ubiquitously expressed in all ATLL cells later than Tax, with increasing levels over time.11,20HBZ has an important role in viral replication, and proliferation, maintenance and immune evasion of ATLL-cells. Thus, HBZ transcribes viral mRNA uniformly expressed in ATLL cells; attenuates host immune responses to avoid destroying ATLL-cells, and shapes a microenvironment favorable to HTLV-1.11,21
Therefore, HBZ antagonizes and complement many of the activities of Tax. Tax acts early while HBZ does late during virus infection. The former is used to initiate transformation (oncogenic effect), and the latter is used to maintain the transformed phenotype of ATLL cells19(Fig. 1).
Although HTLV-1 infects T-cells, B-cells, fibroblasts, dendritic cells, and macrophages, only regulatory T-cells expressing CD25 and the transcription factor forkhead box P3 (FoxP3) are considered to be the cell type that transforms to ATLL.5 FoxP3 expression is important because it exerts both beneficial and harmful effects: it suppresses the growth of autologous ATLL clones but also suppresses the host's cytotoxic T lymphocyte response, which normally limits HTLV-1 replication and reduces the risk of ATLL.11,22
ClassificationATLL was sub-classified in four variants by the World Health Organization Classification of Tumors of Hematopoietic and Lymphoid Tissues in 2008, according to the Shimoyama classification based on organ involvement, lactate dehydrogenase (LDH) and calcium values. The variants are acute (60%) lymphomatous (20%), chronic (15%) and smoldering (5%).23,24
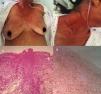
The acute variant is the most aggressive, characterized by marked leukocytosis with atypical lymphocytes, skin lesions (erythematous rash, nodules or papules), constitutional symptoms, massive lymphadenopathy that spare mediastinum and hepatosplenomegaly.25 Hypercalcemia and elevated LDH are frequently present. Respiratory complications may appear due to tumor infiltration or opportunistic infections and sepsis5 (Fig. 2).
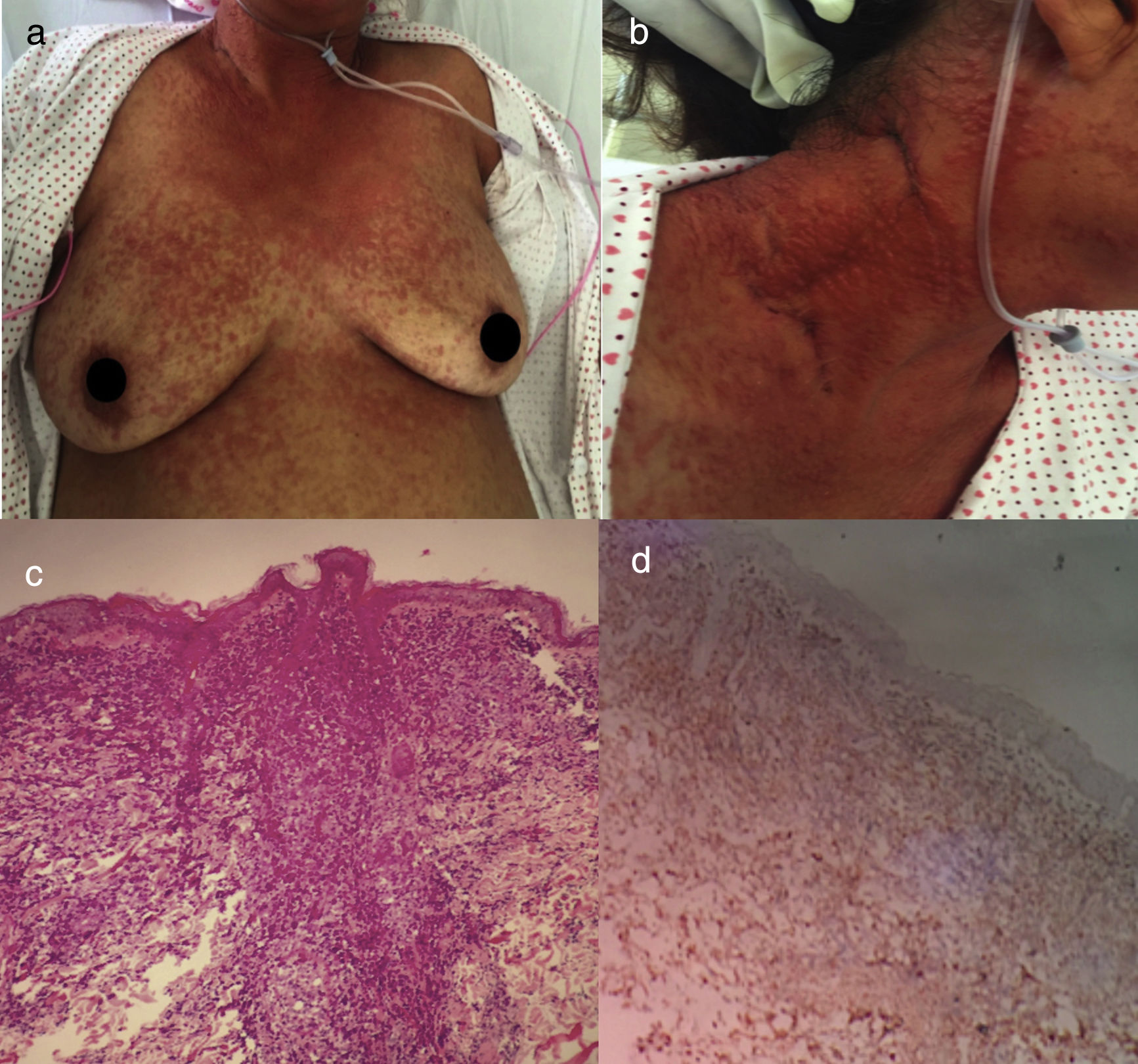
Acute ATLL. (a) 64yo female with multiple papules distributed on trunk and proximal extremities for over three months. (b) Papules are asymptomatic, erythematous, with increased consistency. (c) Infiltration of dermis by atypical lymphoid cells with epidermotropism. Hematoxylin and Eosin stain, x20. (d) Immunohistochemistry CD3+, x20. The patient died after 2 weeks of admission from septic shock.
Lymphomatous variant is also aggressive, but its prominent feature is marked lymphadenopathy without leukemia. Skin involvement and hypercalcemia are less frequent.25,26
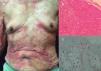
The chronic variant typically presents with an exfoliative skin rash, less prominent leukocytosis with absolute lymphocytosis, mild lymphadenopathy, and hypercalcemia25 (Figs. 3 and 4).
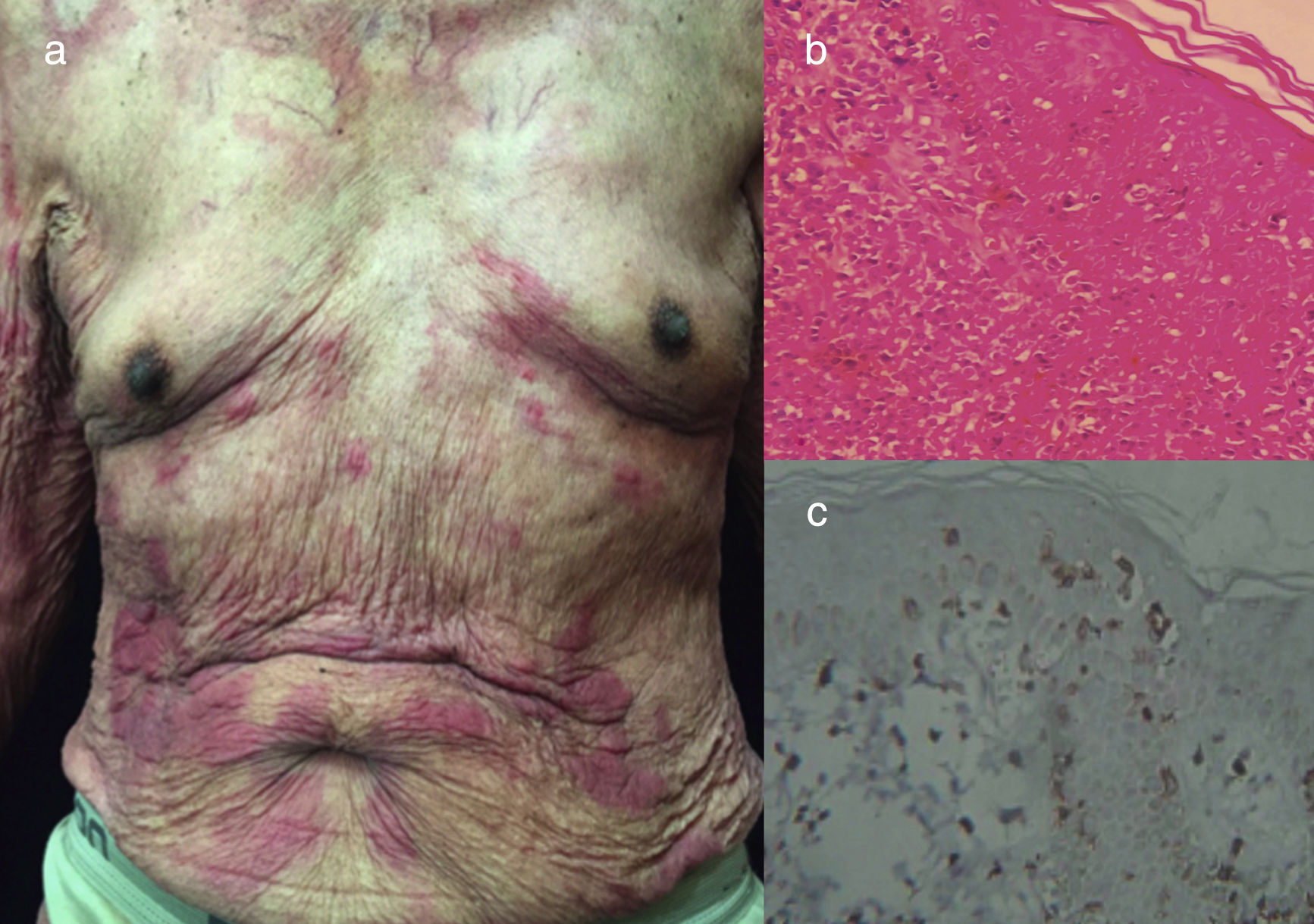
Chronic ATLL. (a) 91yo male with multiple pruritic and erythematous plaques in trunk for over a year, associated with severe xerosis. (b) Epidermotropism and Pautriers abscess. Hematoxylin and Eosin stain, x40. (c) Immunohistochemistry CD3+, x40. The patient died after 4 months of admission. He was under support care and symptomatic treatment.
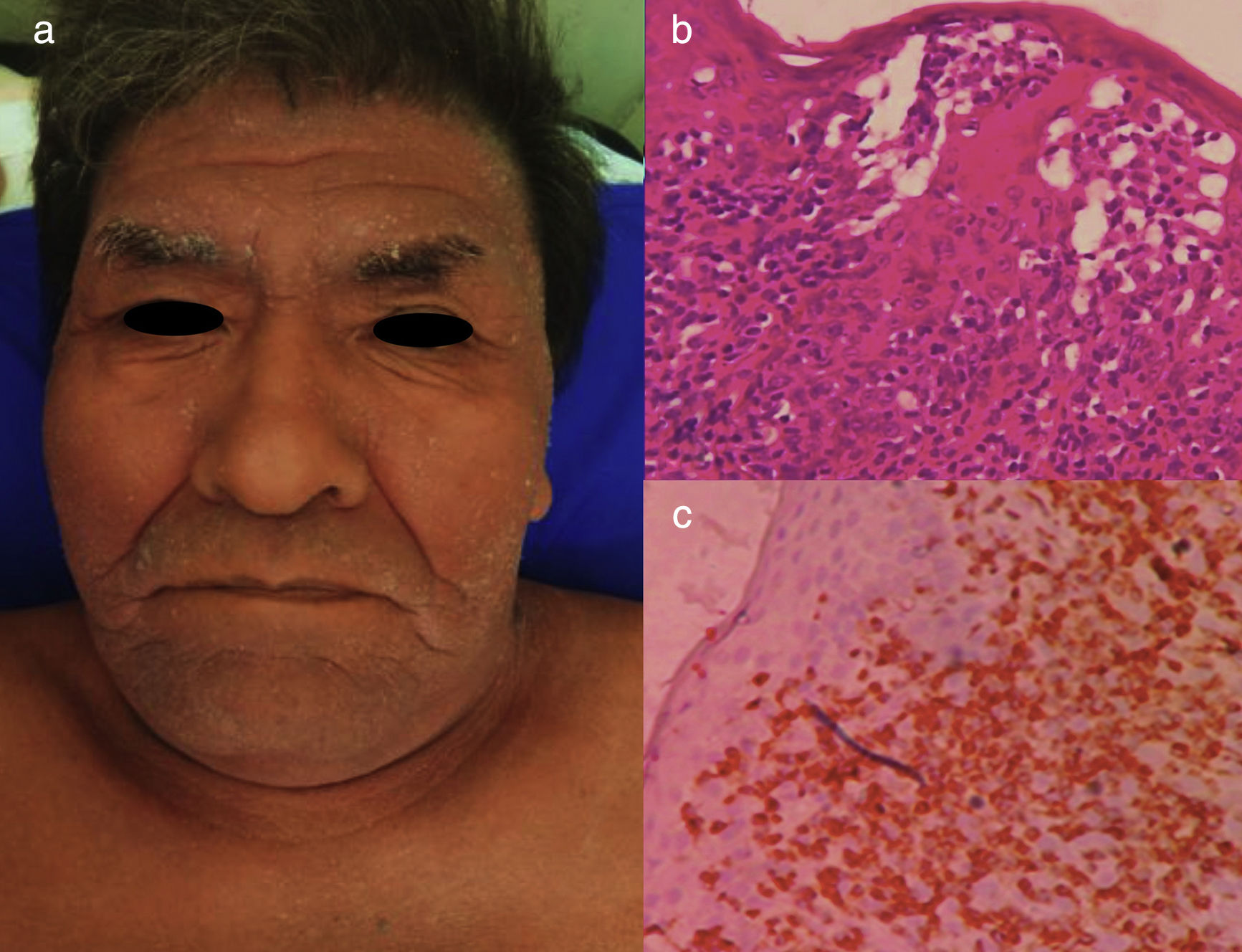
Chronic ATLL. a) 66yo male with erythroderma and severe xerosis for over 3 years. (b) Epidermotropism and Pautriers microabscess. Hematoxylin and Eosin stain, x40. (c) Immunohistochemistry CD3+, x40. The patient died after 8 months of chemotherapy with gemcitabine (1000mg/m2 per day) and oxaliplatin (100mg/m2 per day) every 3 weeks.
The smoldering variant usually is asymptomatic. Patients have a normal WBC with <5% circulating neoplastic cells and there is not hypercalcemia or organomegaly associated. Similar to the chronic variant, the smoldering type has a 25% risk of transforming to the acute phase.5
Clinical findingsCutaneous lesions are present in 43% to 72% of all clinical variants of ATLL, due to direct invasion of malignant cells to skin, resulting in diverse type of eruptions. There are six types of skin eruptions: nodulotumoral (38.7%), plaque (26.9%%), multipapular (19.3%), patch (6.7%), erythrodermic (4.2%), and purpuric (4.2%) Skin eruptions are not exclusive and are frequently chronic, painless or pruritic.27,28 Generalized eruption is present in 70%, isolated trunk involvement in 10%, the involvement of trunk and limbs in 25%, and isolated involvement of the head, neck or limbs in 5%29 (Fig. 2 to 4).
There have been reports of rare forms of cutaneous involvement of ATLL such as dyshidrotic eczema-like lesions of the hand and foot, keloid-like lesions, granuloma-like lesions, and a case with vesicular and bullous lesions.30 In addition, other forms of presentation described were eczematous-like, sclerodermatous-like and ichthyosiform-like lesions.31
Of note, the type of ATLL skin eruption present has been identified as an independent prognostic factor. Patches and plaques have been associated with better survival whereas erythrodermic and nodulo-tumoral presentations have the poorest prognosis.28
Laboratorial findings and diagnosisHistology shows 3 different patterns of infiltration: perivascular, nodular, and diffuse. Epidermotropism and Pautrier microabscess are common features32 (Fig. 2 to 4). As those findings may resemble other cutaneous T-cell lymphomas, IHC plays a central role in the differential. ATLL cells express mature T-cell markers, like CD2, CD5, CD25 (strong and uniform), CD29, CD45RO, TCR, and HLA-DR; and also they commonly express CD4+ and CD8-.33 CD25 expression (IL-2 receptor) may be distinctive but not specific, as it can also be present in Pro-T-lymphocytic leukemia and Sézary syndrome (SS).33
Peripheral blood involvement is detected in the acute and chronic variants, mainly with leukocytosis with lymphocytosis. Peripheral blood smear demonstrates blasts known as “flower cells” that are medium- to large-sized lymphocytes with petal-like nuclei and coarse chromatin, CD4+ and CD25+.34 They are frequently confused with Gumprecht shadows.
BM biopsy usually shows subtle or patchy infiltrate of atypical lymphoid cells with irregular nuclear contours (Fig. 2). However, it is not imperative for ATLL diagnosis.35 Also, BM involvement of >5% of ATLL cells on aspirate or biopsy is considered an independent poor prognostic factor.5 Involved lymph nodes typically exhibit diffuse involvement, with primarily paracortical expansion.36
Flow-cytometric analysis of T-cells in ATLL shows that this is a malignancy of regulatory T-cells/Th2 cells with high-frequency expression of CD3/CD4/CD25/CCR4 and FoxP3 in about half of the cells.
ATLL diagnosis is based on a combination of clinical features, morphologic and immunophenotypic changes of the malignant cells, along with confirmation of HTLV-I infection, by tests for specific antibodies and/or polymerase chain reaction (PCR) in peripheral blood cells,.37,35 Identification of at least 5 percent tumor cells by cytology and immunophenotype in the peripheral blood with confirmation of HTLV-1 infection is often sufficient to make the diagnosis in patients with acute, chronic, or smoldering ATLL types. Lymphomatous variant needs an excisional biopsy of an involved lymph node for histopathologic examination.38 Laboratory for clonal proviral integration is not routinely performed; therefore diagnosis is usually confirmed.
The pathologic differential diagnosis for ATLL includes peripheral T-cell lymphoma unspecified (PTCLu), anaplastic large cell lymphoma, cutaneous T-cell lymphomas such as mycosis fungoides (MF) and SS, and angioimmunoblastic T-cell lymphoma. PTCLu is the most common T-cell lymphoma in North America and may be pathologically indistinguishable from ATLL. Also, MF and SS are important differentials to consider, as similar clinical findings and histologic infiltration of lymphocytes are seen. If a history of migration from an endemic region is not elicited, the diagnosis of ATLL may be overlooked. Misdiagnosis is very easy in low prevalence areas, but positive HTLV-1 serology or confirmation of viral integration in neoplastic cells will favor ATLL in all cases.39
Prognosis and Outcome predictorsIn general, the ATLL prognosis is worse than other PTCLu. The aggressive variants, acute and lymphomatous, have the worst prognoses, with a median survival time (MST) of 6 months to 10 months, respectively. Meanwhile, the indolent variants such as chronic and smoldering have the longest MST, near 2 years and over 2 years, respectively.25,36 Death is most often caused by infectious complications, uncontrolled hypercalcemia or progressive disease.36
Several prognostic systems have been developed to identify groups with a significantly better prognosis. 807 patients were grouped according to prognostic factors such as the Ann Arbor stage (I and II vs III and IV), performance status (PS) (0 to 1 vs 2 to 4), age, serum albumin, and soluble IL-2 receptor (CD25). Authors estimated a linear risk function called ATLL-PI based on Cox regression coefficients as follows: ATLL-PI = 0.65 (if stage = III or IV) + 0.35 (if PS > 1) + 0.016 X age (years) - 0.36 X albumin (g/dL) + 0.37 X log10 (IL-2 receptor [U/mL]). The low-risk group was defined as an ATLL-PI < 1.6; intermediate-risk group as 1.6 ≤ ATLL-PI > 2.6; and high-risk group as ATLL-PI ≥ 2.6. The MST was 3.6, 7.3 and 16.2 mo., in the high, intermediate and low-risk groups, respectively.40
The Japan Clinical Oncology Group (JCOG) Lymphoma Study Group (LSG) came up with two significant prognostic factors: PS and hypercalcemia. Moderate-risk group (corrected calcium <2 75 mmol/l and a PS of 0 or 1) have an MST and 5-year overall survival (OS) of 14 months and 18%, respectively. The high-risk group (corrected calcium ≥2 75mmol/l and/or a PS of 2–4) have an MST and 5-year-OS of 8 months and 4%, respectively.41
As different variants would be associated with distinct prognostic factors, a study with 95 ATLL patients in Peru found useful scores to risk-stratify patients with aggressive ATLL.42 Low albumin level and the presence of B symptoms were independent factors for worse survival in lymphomatous ATLL, and high beta2-microglobulin level was an independent factor for worse survival in acute ATLL.42
Of note, some prognostic factors can also dictate treatment decisions. Here we found clinical factors as PS, LDH, age, stage, number of lesions, and hypercalcemia; and also molecular factors, such as Ki-67 expression, soluble IL-2 receptor, alteration of p53, and overexpression of interferon regulatory factor-4 (IRF-4).11
TreatmentIn general terms, studies have shown excellent survival measures with zidovudine/interferon α (AZT/IFN) treatment in patients with indolent ATLL, and multidrug chemotherapy and/or allogeneic stem cell transplantation (Allo-HSCT) for aggressive forms in Japan. In Western countries, AZT/IFN with or without chemotherapy is the treatment of choice.5
The recent chemotherapy regimes, such as the modified LSG15 regimen, consists of six cycles of vincristine, cyclophosphamide, doxorubicin, and prednisone (VCAP); doxorubicin, ranimustine, and prednisone (AMP); and vindesine, etoposide, carboplatin, and prednisone (VECP). This regimen showed a 3-year survival rate of 24%, a complete response rate of 40%, and an MST of 13 months in 118 previously untreated patients with aggressive ATLL.43
On the other hand, IFN/AZT therapy may be promising, especially for types with leukemic manifestation.11 A recent meta-analysis showed a 5-year OS rate of 46% for 75 patients who received first-line antiviral therapy, 20% of 77 patients who received first-line chemotherapy, and 12% of 55 patients who received first-line chemotherapy followed by antiviral therapy.44 Of note, the dosing of interferon and antiviral has not been standardized yet. One approach is to initiate INFα 2b at a dose of 5MU subcutaneously daily for 8 to 12 weeks in combination with AZT orally 300mg tid. On completion of IFNα therapy, AZT at 300mg three times daily is continued indefinitely.45
Allo-HSCT is promising for the treatment of aggressive ATLL, possibly reflecting graft-versus-ATLL effect. In a comparative study of 56 patients, those who received chemotherapy after allo-HSCT or only chemotherapy had a 3-year OS of 44.9% and 27.7%, respectively.46,47 Some authors propose that allo-HSCT should be considered early, before 100 days of ATLL diagnosis. In 72 patients with ATLL, those who received early vs late allo-HSCT had a 4-year OS of 49% and 31%, respectively.48
The International Consensus Meeting of the American Society of Clinical Oncology published the current guidelines for ATLL in 200935 (Fig. 5). First, smoldering and favorable chronic ATLL variants, when asymptomatic, should be under observation. Symptomatic variants (e.g. skin lesions, opportunistic infections) should consider AZT/IFN or observation. Unfavorable chronic and acute variants with good prognostic factors should consider chemotherapy with VCAP-AMP-VECP or AZT/IFN. Acute variants with poor prognostic factors should consider chemotherapy followed by conventional or reduced-intensity allo-HSCT.
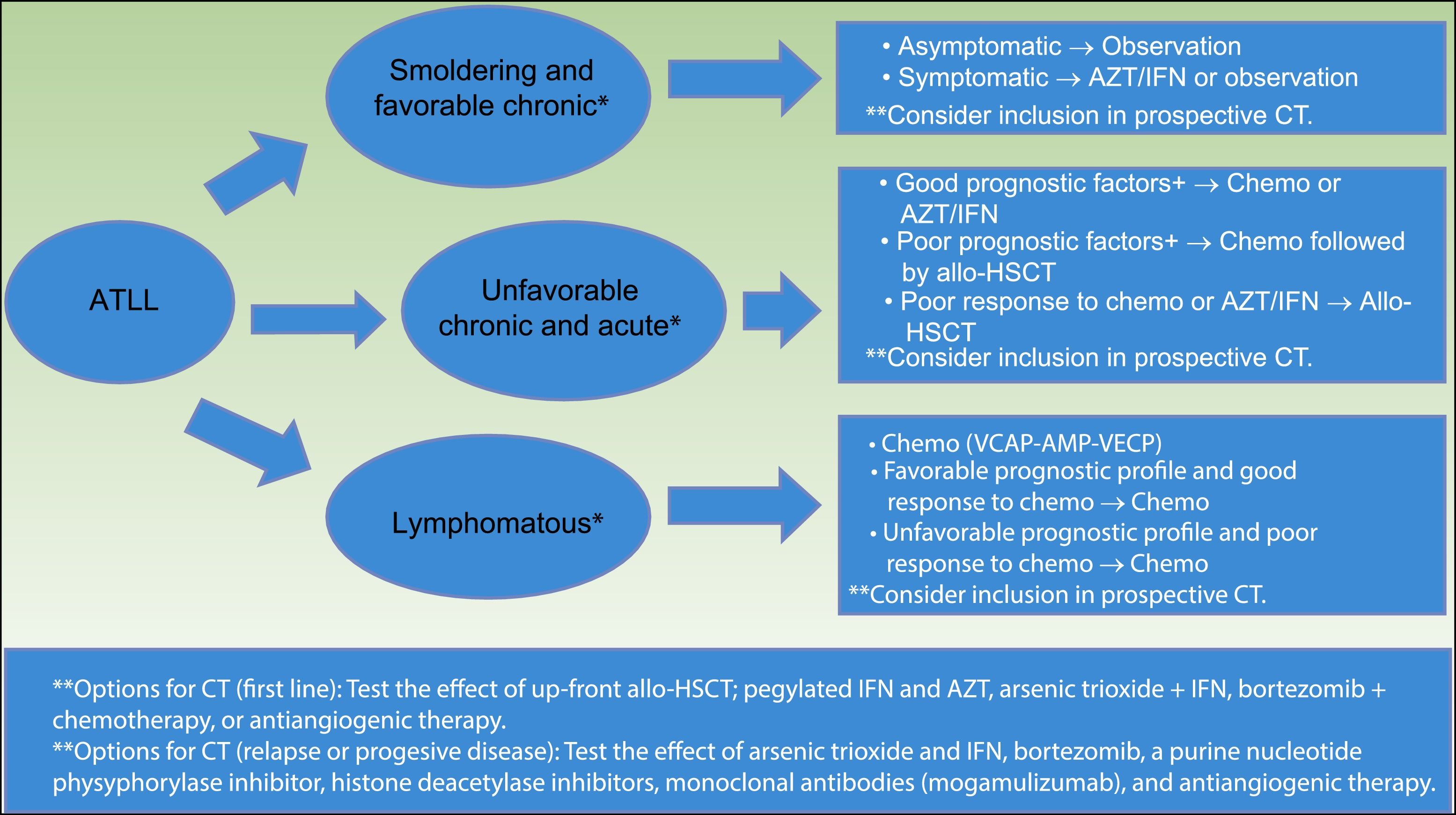
Strategy for the treatment of ATLL, proposed by the American Society of Clinical Oncology in 2009(35). *Favorable and unfavorable disease is based on prognostic factors that include clinical factors, such as performance status, LDH, age, stage, the number of involved lesions, and hypercalcemia; and molecular factors, such as Ki-67 expression, soluble IL2 receptor, alteration of p53, and overexpression of IRF-4. **Clinical trials options include testing the effect of allo-HSCT, combined therapy with arsenic trioxide, IFN, bortezomib, VCAP-AMP-VECP, or antiangiogenic therapy; and testing pegylated IFNα and AZT, monoclonal antibodies (Mogamulizumab).
Abbreviations ATLL: adult T-cell leukemia–lymphoma; AZT/IFN: zidovudine/interferon α; CT: clinical trials; Chemo: chemotherapy; allo-HSCT: allogeneic stem cell transplantation; VCAP: vincristine cyclophosphamide doxorubicin and prednisone; AMP: doxorubicinranimustineand prednisone; VECP: vindesine etoposide carboplatinand prednisone.
Patients with poor response to initial therapy with chemotherapy or AZT/IFN should consider conventional or reduced-intensity allo-HSCT.35
Lymphomatous variant should consider chemo with VCAP-AMP-VECP. If favorable prognostic profiles and good response to initial therapy are present, patients should keep on chemotherapy. If unfavorable prognostic profiles or poor response to initial therapy, patients should consider conventional or reduced intensity allo-HSCT.35
CCR4 is expressed on the neoplastic cells of most patients with ATLL, and this expression has been associated with cutaneous manifestation and poor prognosis.11 The defucosylated humanized anti-CCR4 antibody Mogamulizumab (Moga) has been recently approved in Japan for the treatment of refractory or relapsed ATLL. Moga exhibits very strong cytotoxicity for ATLL cells via antibody-dependent cellular cytotoxicity and depletes regulatory T-cells for at least a few months.49 Therefore, major concerns have risen about the harmful influences of pretransplant Moga as it may increase the risk of graft-versus-host disease after the allo-HSCT.50 In addition, serious adverse reactions such as Stevens-Johnson syndrome have been reported.51 Moga has a response rate of approximately 50% in relapsed/refractory ATLL.52 Nevertheless, recent trials have proven the superiority of the combination of Moga with modified LSG15 versus chemotherapy alone for untreated aggressive ATLL. Combination therapy produced a higher complete response rate of 52% vs 33%, respectively.53
ConclusionATLL is an aggressive neoplasm of regulatory T-cells, caused by chronic infection of HLTV-1, with a difficult diagnosis, and poor prognosis. As HTLV-1 is endemic in several native groups in Japan and Latin America, dermatologists should consider ATLL diagnosis in patients with compatible skin eruptions and epidermotropism with a lymphocytic dermal invasion on histology. Clinical and laboratory findings define ATLL subtypes, which relates to prognosis. MF and SS are the main differentials, with similar clinical and histological findings. While indolent variants may benefit from AZT/INF regimes, aggressive variants may respond to chemotherapy and allo-HSCT. The monoclonal antibody Mogalizumab has been approved for refractory or relapsed cases. Dermatologists need to see the dimension of this complex disease, as the HTLV-1 infection is still a public health challenge, but few resources and scarce research are advocated to resolve this problem in Latino countries.
We thank Dra. Dina Carayhua from the Dermatopathology Department for her contribution in the analysis and collection of histopathology cases.
Please cite this article as: Rodríguez-Zúñiga MJM, Cortez-Franco F, Qujiano-Gomero E. Leucemia/linfoma de células T del adulto. Revisión de la literatura científica. Actas Dermosifiliogr. 2018;109:399–407.


