



HISTORIA CLINICA
Una mujer de 27 años de edad, sin antecedentes personales ni familiares de interés, consultó por presentar lesiones en el dorso de las manos ante mínimos traumatismos desde hacía 1 año. No refería ingesta de medicamentos ni presentaba sintomatología sistémica asociada.
EXPLORACION FISICA
En la exploración cutánea se observaban pequeñas vesículas de contenido seroso en el dorso de las manos así como cicatrices atróficas y quistes de milio (fig. 1). El resto de la exploración fue anodina.
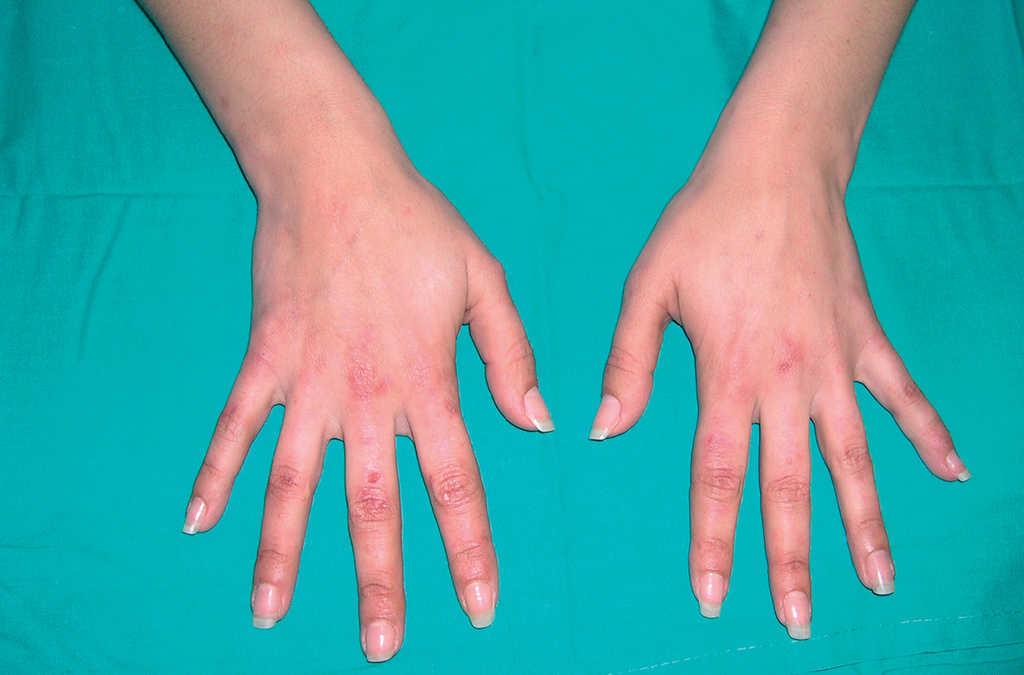
Fig. 1.--Pequeñas vesículas de contenido seroso, quistes de milio y cicatrices atróficas en dorso de manos.
EXPLORACIONES COMPLEMENTARIAS
Los estudios complementarios que incluían hematimetría, pruebas de función hepática y renal, serología de virus de la hepatitis B y C (VHB y VHC), niveles de hierro y de porfirinas en orina y heces, revelaron los siguientes resultados patológicos: uroporfirinas 54 μg/24 h (valores normales [VN], hasta 22 μg/24 h), coproporfirinas 901 μg/24 h (VN hasta 60 μg/24 h), porfobilinógeno 4,6 mg /24 h (VN hasta 2,0 mg/24 h) en orina de 24 h, protoporfirinas 72,4 μg/g (VN hasta 8,0 μg/g) y coproporfirinas 44,3 μg/g (VN hasta 5,0 μg/g) en heces. Se realizó además una biopsia cutánea (figs. 2 y 3).
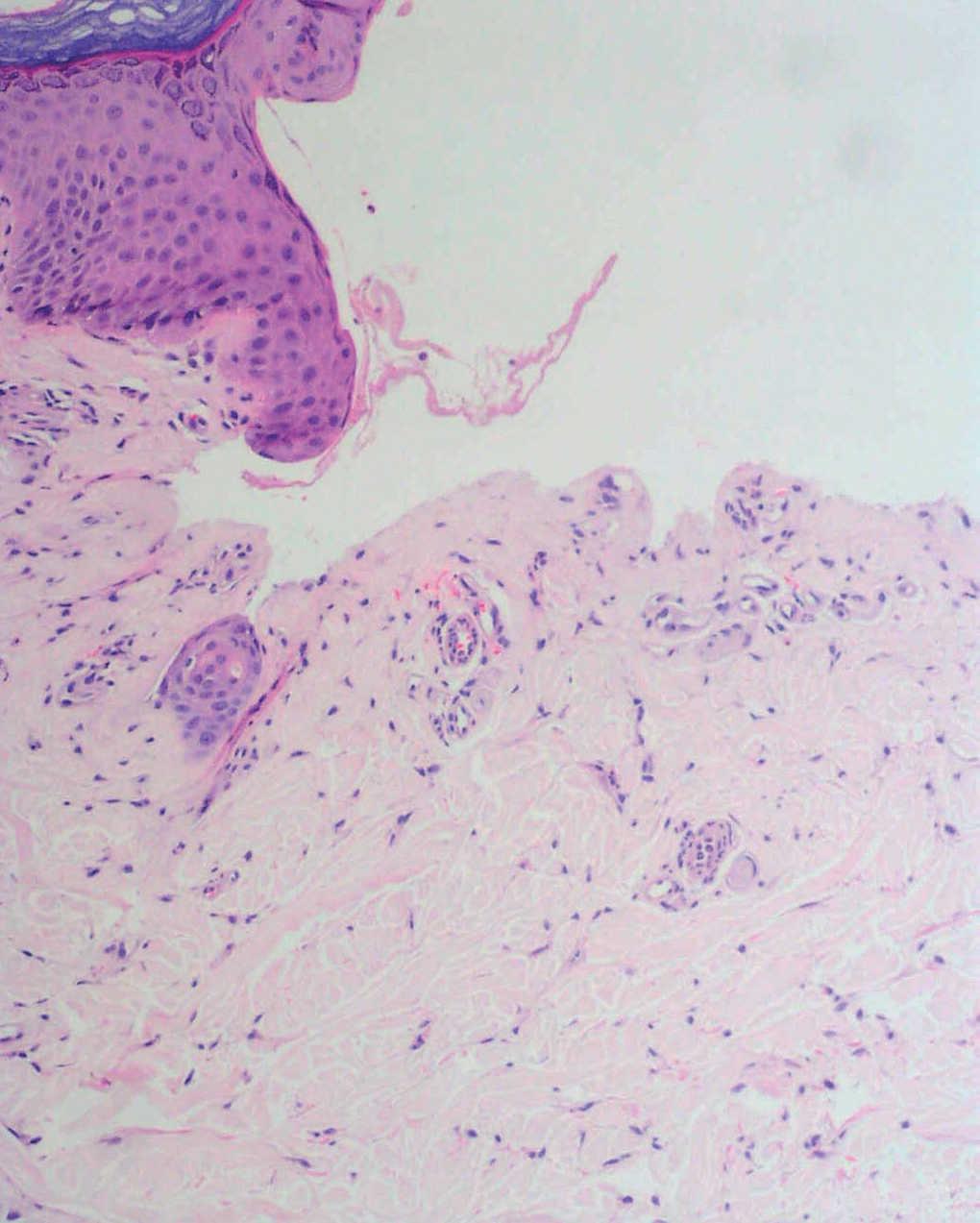
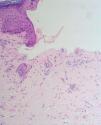
Fig. 2.--Ampolla subepidérmica sin infiltrado inflamatorio y con aspecto festoneado de las papilas dérmicas.

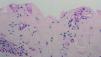
Fig. 3.--Depósitos perivasculares con la tinción del PAS.
DIAGNOSTICO
Porfiria variegata.
HISTOPATOLOGIA
El estudio histopatológico mostró una ampolla subepidérmica con ausencia de infiltrado inflamatorio. La técnica de PAS reveló la presencia de anillos hialinos perivasculares.
EVOLUCION
Se inició tratamiento con β -carotenos y fotoprotectores, observándose una discreta mejoría de las lesiones cutáneas. Hasta el momento actual la paciente no ha presentado sintomatología neurovisceral.
COMENTARIO
La porfiria variegata es una forma rara de porfiria hepática aguda que se hereda con carácter autosómico dominante. Se debe al déficit enzimático de protoporfirinógeno oxidasa (PPOX) cuyo gen se ha localizado en el cromosoma 1q22-23 y del que hasta la fecha se han descrito más de 80 mutaciones 1. La mayor prevalencia se recoge en Sudáfrica (1/300) y Finlandia (2/100.000).
La mayoría de los individuos que heredan el gen permanecen asintomáticos (latentes) a lo largo de su vida, especialmente si evitan los factores precipitantes. Clínicamente la sintomatología es variada e incluye ataques neuroviscerales agudos similares a la porfiria aguda intermitente (PAI) y lesiones de fotosensibilidad cutánea indistinguibles de la porfiria cutánea tarda (PCT) 2. Estos síntomas no suelen aparecer hasta la pubertad. En las mujeres suele manifestarse con síntomas neuroviscerales, mientras que los varones lo hacen con lesiones cutáneas. Sólo entre el 30 y el 45 % de los pacientes desarrollan lesiones cutáneas. La fotosensibilidad comienza entre la segunda y tercera décadas. Después de la exposición solar aparecen vesículas en cara, cuello y dorso de las manos que pronto se tornan hemorrágicas. Las ampollas también aparecen ante pequeños traumatismos debido a la fragilidad cutánea. La curación cursa con la formación de cicatrices en papel de fumar y quistes de milio. La cara toma un aspecto curtido con exageración de los surcos de la frente, hipertricosis, hiper e hipopigmentación. Los síntomas neuroviscerales incluyen estados confusionales, dolor abdominal y neuropatía periférica. Estos síntomas pueden precipitarse por múltiples factores entre los que se incluyen determinados fármacos, el embarazo, el ciclo menstrual, el alcohol y la reducción en la ingesta de hidratos de carbono 3.
El diagnóstico se apoya en los hallazgos clínicos, el patrón de excreción de porfirinas en orina y heces y en la demostración de un pico de fluorescencia en plasma a 626 nm mediante espectrofluorimetría 4.
Es característica la elevación de porfobilinógeno (PFB) y ácido aminolevulínico (ALA) en orina durante los ataques, que se mantienen elevados en el 50 % de los casos. En la orina de individuos asintomáticos existe una elevación de las uroporfirinas y de las coproporfirinas con predominio de estas últimas (copro > uro). Del mismo modo en heces existe un incremento de protoporfirinas y coproporfirinas con un claro predominio de protoporfirinas (proto > copro).
El estudio histopatológico recuerda a una PCT con una ampolla subepidérmica y con un escaso infiltrado inflamatorio. Los vasos de la dermis muestran depósitos de un material PAS positivo. Con microscopia electrónica se observa una reduplicación de la membrana basal y un depósito perivascular de un material fibrilar fino 5.
Esta entidad plantea diagnóstico diferencial con la porfiria aguda intermitente (PAI) y la porfiria cutánea tarda (PCT). En la PAI no existen lesiones cutáneas ya que los precursores de las porfirinas que se acumulan (ALA y PBG) no son fotosensibilizantes. Además no hay elevación de porfirinas en heces.
La PCT no cursa con síntomas neuroviscerales y el patrón de excreción de porfirinas en orina es uro > copro, con valores de PFB normales.
La prevención de los síntomas neuroviscerales es de extrema importancia; por ello, los pacientes deben evitar los fármacos y factores precipitantes. Unos niveles de coproporfirinas en orina superiores a 1.000 nmol/día se relacionan con un mayor riesgo de presentar tanto lesiones cutáneas como síntomas neuroviscerales 6.
Respecto al manejo de las lesiones cutáneas se debe evitar el sol y usar pantallas solares. Las cantaxantinas y los β -carotenos parecen aportar algún beneficio, aunque se han descrito casos de retinopatía cristalina por éstos.
Debe realizarse un cribado de los familiares de primer y segundo grado. La espectrofluorimetría en suero sería la técnica más sensible, sobre todo en prepúberes en los que la concentración de porfirinas fecales no está elevada. Esta prueba tiene una sensibilidad del 60-100 %, es rápida y barata en contraste con el análisis de porfirinas en heces, que tiene una sensibilidad del 35 %. Hoy día, la mayoría de los estudios se centran en el estudio de posibles mutaciones del gen cuya sensibilidad es del 100 %.


