











La piel es el órgano más extenso y más expuesto del cuerpo humano. Ello implica un magnífico terreno para el diagnóstico precoz de las enfermedades sistémicas que cursan con afectación sistémica y para las cuales la piel se vuelve un marcador diagnóstico. Las bases conceptuales y los criterios diagnósticos de muchas de estas entidades se han visto modificados o ampliados en los últimos años, con lo que la aproximación a la biopsia cutánea y la evaluación de los signos dermatopatológicos útiles en el diagnóstico precoz han variado también. En esta revisión intentamos hacer un enfoque de algunos de los procesos sistémicos con repercusión cutánea que más han variado conceptualmente en las últimas décadas.
The skin is the largest and most exposed organ in the human body and the ideal place to look for signs that aid in the early diagnosis of systemic diseases with cutaneous effects. As the concepts that underpin our understanding of many of these diseases have evolved or expanded in recent years, there have also been changes in the criteria we use for early diagnosis, including our approaches to skin biopsy and dermatopathologic evaluation. This review focuses on some of the systemic processes with skin manifestations for which our basic understanding has changed most in recent decades.
La piel es el órgano más extenso y más expuesto del cuerpo y, en consecuencia, el primero en el que se reflejan muchas afectaciones sistémicas.
Una revisión de todos los cuadros sistémicos que tienen un reflejo cutáneo sería imposible incluso en un capítulo de libro. En el presente artículo nos centraremos en algunos aspectos recientes o novedosos sobre algunas de las enfermedades multiorgánicas que tienen traducción dermatológica, así como en el abordaje de las que más se han perfilado en los últimos años, habiendo adquirido un nuevo enfoque conceptual para muchas de ellas.
Enfoque histopatológico de las enfermedades autoinflamatoriasLas enfermedades autoinflamatorias (EAI) son trastornos crónicos heterogéneos resultado de un fenotipo anormal en la inmunidad innata1. Esto resulta en un defecto de los inflamasomas (oligómeros multiproteicos de las células mieloides) que provoca una respuesta inflamatoria excesiva, regulada por interleucina-1, ante un grupo de agentes activadores diversos.
Teóricamente se trata de trastornos no infecciosos, no alérgicos, no autoinmunes y no inmunodeficientes. Sin embargo, el enfoque actual es que la mayor parte de los trastornos autoinmunes y autoinflamatorios son en realidad entidades mixtas en un espectro continuo.
Originalmente se distinguían tres tipos principales de EAI: el síndrome de Sweet (dermatosis febril neutrofílica aséptica), el pioderma gangrenoso y la hidradenitis supurada. En las últimas décadas se han añadido varias entidades asociadas a enfermedades vasculares, mutaciones genéticas determinadas, vasculitis, mielodiscrasias, trastornos hereditarios o tumores sólidos. Entre las que ahora se consideran EAI están el eritema elevatum diutinum, los síndromes de dermatosis/artritis asociados a enfermedad intestinal, los cuadros erisipeloides asociados a fiebre mediterránea familiar, las erupciones urticariales y los síndromes periódicos asociados a criopirina o al receptor del factor de necrosis tumoral.
Dos de las entidades que han ido «recolocándose» en el terreno de las EAI son la dermatitis granulomatosa intersticial y la dermatitis granulomatosa neutrofílica en empalizada, entendidas mejor en el momento actual como los polos de un espectro morfológico continuo2.
Recapitulando, las EAI se distribuyen en un amplio espectro. En los polos estarían, de un lado, las raras enfermedades autoinflamatorias puras monogénicas (DITRA, DIRA, síndrome de Blau, FMF, TRAPS, HIDS, PAPA…); del otro, las raras enfermedades autoinmunes monogénicas (ALPS, IPEX, APCED…), y en medio, todo el espectro de patologías que entremezclan componente autoinmune y autoinflamatorio con predominio de uno u otro. Estarían en este último grupo: a)las enfermedades autoinmunes poligénicas clásicas (artritis reumatoide, anemia perniciosa, tiroiditis autoinmune, enfermedad de Addison, miastenia gravis, vasculitis con ANCA, lupus eritematoso sistémico, síndrome de Sjögren, diabetes mellitus tipo1, enfermedad celiaca…); b)las EAI poligénicas clásicas (enfermedad de Crohn, colitis ulcerosa, gota, artritis reactiva sin relación con el complejo mayor de histocompatibilidad, vasculitis sin asociación con anticuerpos, uveítis idiopática…), y c)un grupo mixto con componentes autoinmune y autoinflamatorio por igual (artritis reactiva, espondilitis anquilosante, artritis psoriásica, síndrome de Behçet, uveítis asociada a HLA-B27…).
En el grupo de EAI monogénicas, varias entidades nuevas han sido introducidas en los últimos años, como el PAPA (artritis piogénica, pioderma gangrenoso y acné), el PAPASH3 (artritis piogénica, pioderma gangrenoso, acné e hidradenitis supurada), el PASH (pioderma gangrenoso, acné, hidradenitis supurativa4 o el PsPASH (artritis psoriásica, pioderma gangrenoso, acné e hidradenitis supurada).
Las manifestaciones cutáneas en las EAI son frecuentes, y en muchas ocasiones una pieza fundamental del diagnóstico. Entre ellas, las erupciones urticariales o maculopapulares, las pústulas, las aftas, el pioderma gangrenoso, las lesiones granulomatosas y las de tipo erisipela. Aunque ninguna es patognomónica de un síndrome concreto, todas tienen en común el infiltrado neutrofílico.
La inmunidad innata está gobernada por neutrófilos, basófilos y eosinófilos, con lo que no es de extrañar que el «marcador histopatológico» principal de las EAI sea el polimorfonuclear neutrófilo, con o sin vasculitis. Aunque esto es muchas veces una obviedad en la preparación de rutina con hematoxilina-eosina, en ocasiones se requieren técnicas especiales, como mieloperoxidasa, para demostrarlo5. Ello es así porque el componente neutrofílico es frágil y puede aparecer aplastado, fragmentado y enmascarado por células más resistentes como los linfocitos, simulando un infiltrado crónico en lugar de agudo.
Algunos estudios apuntan a que los infiltrados neutrofílicos de las EAI son clonales o pseudo-clonales6, aunque dicha línea de investigación ha sido controvertida en años posteriores.
Los infiltrados neutrofílicos pueden encontrase en distintos estratos de la piel dependiendo de la entidad. Así, son preferentemente epidérmicos en la psoriasis pustulosa, en la pustulosis exantemática generalizada (fig. 1A), en la queratodermia blenorrágica, en la dermatosis pustular subcórnea (Sneddon-Wilkinson), en el pénfigo IgA, en la pustulosis amicrobiana de los pliegues, en la acropustulosis infantil o en la pustulosis neonatal transitoria. Por el contrario, el infiltrado neutrofílico es fundamentalmente dérmico en entidades como el síndrome de Sweet, el pioderma gangrenoso, la enfermedad de Behçet, el síndrome artritis-dermatosis asociado a enfermedad intestinal, la hidradenitis ecrina neutrofílica, la dermatitis neutrofílica reumatoidea, la urticaria neutrofílica, la enfermedad de Still, el eritema marginado o el síndrome de fiebre periódica hereditaria. Incluso en este último grupo, tanto la densidad como la profundidad del infiltrado dérmico son variables: más superficial en el Sweet —por ejemplo, figura 1B,C)—, con presentaciones más profundas en el Behçet o infiltrando toda la dermis y penetrando en la hipodermis en el pioderma gangrenoso.
Infiltrado neutrofílico preferentemente epidérmico en la pustulosis exantemática generalizada (H&E ×100). B)Infiltado difuso dérmico rico en polimorfonucleares neutrófilos maduros con intenso edema subdérmico y áreas de hemorragia en dermis papilar (H&E ×20). C)A este aumento, los polimorfonucleares neutrófilos, así como la cariorrexis, se ven con mayor detalle (H&E ×200).")
A)Infiltrado neutrofílico preferentemente epidérmico en la pustulosis exantemática generalizada (H&E ×100). B)Infiltado difuso dérmico rico en polimorfonucleares neutrófilos maduros con intenso edema subdérmico y áreas de hemorragia en dermis papilar (H&E ×20). C)A este aumento, los polimorfonucleares neutrófilos, así como la cariorrexis, se ven con mayor detalle (H&E ×200).
La enfermedad de Still es un trastorno inflamatorio sistémico de etiología desconocida que se presenta clínicamente con picos de fiebre intermitente, artralgias, adenopatías generalizadas, dolor de garganta, esplenomegalia y alteración de la función hepática7. A veces el cuadro puede acompañarse de serositis, miopericarditis, enfermedad pulmonar intersticial y afectación neurológica7.
La principal manifestación cutánea en el síndrome de Still es un rash evanescente constituido por una erupción maculopapulosa asalmonada8. Sin embargo, se han descrito9 formas atípicas, acompañadas o no del característico rash. Los síntomas sistémicos pueden ser concomitantes al rash, precedentes en semanas o incluso años, o posteriores hasta en años.
Dentro de las manifestaciones atípicas, lo más frecuente son las pápulas o placas persistentes10-12 (en ocasiones con distribución lineal o reticulada)13, las reacciones de tipo urticarial14,15 o prurigo pigmentosa-like16, el eritema persistente generalizado no pruriginoso o una erupción vesiculopustulosa9.
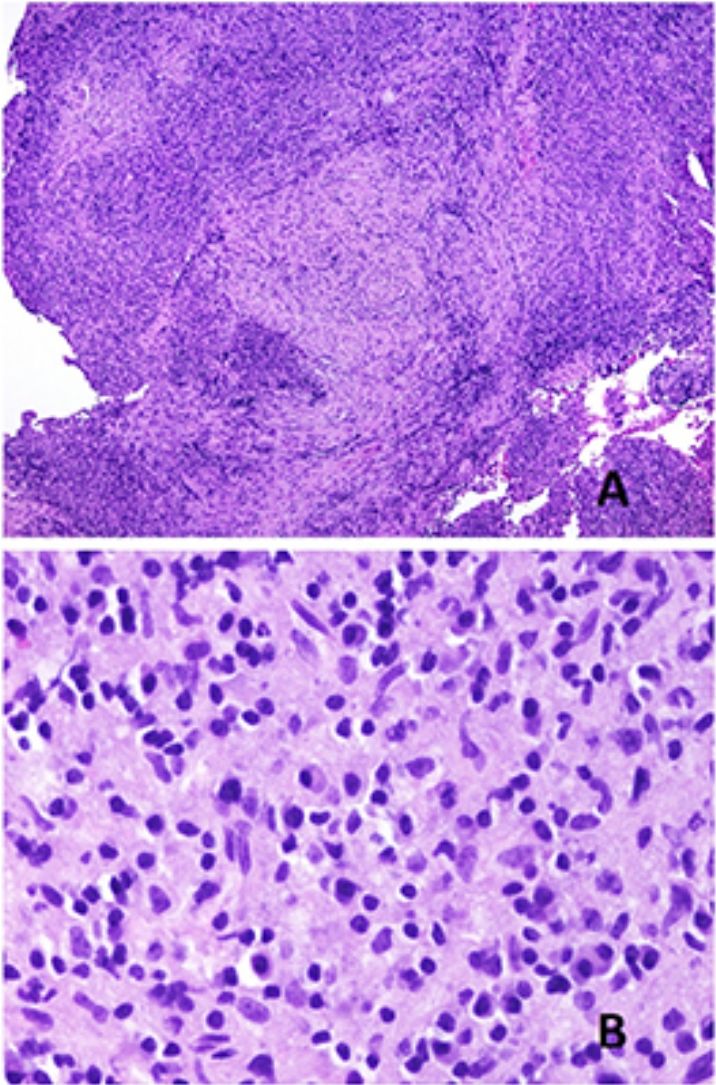
El cuadro histopatológico del rash evanescente clásico de la enfermedad de Still es inespecífico, con un infiltrado superficial que incluye neutrófilos, por encima del cual la epidermis aparece conservada (fig. 2A,B).
Biopsia de un rash evanescente de una enfermedad de Still mostrando un infiltrado inflamatorio perivascular superficial linfohistiocitario (H&E ×100) con algunos neutrófilos (B: H&E ×400). C)En las biopsias de enfermedad de Still crónica sigue siendo evidente un infiltrado inflamatorio perivascular superficial con algunos neutrófilos (H&E ×100), pero se observan además queratinocitos necróticos en las partes altas de la epidermis (D: H&E ×400).")
A)Biopsia de un rash evanescente de una enfermedad de Still mostrando un infiltrado inflamatorio perivascular superficial linfohistiocitario (H&E ×100) con algunos neutrófilos (B: H&E ×400). C)En las biopsias de enfermedad de Still crónica sigue siendo evidente un infiltrado inflamatorio perivascular superficial con algunos neutrófilos (H&E ×100), pero se observan además queratinocitos necróticos en las partes altas de la epidermis (D: H&E ×400).
Por el contrario, en las lesiones persistentes se observan queratinocitos necróticos aislados o bien en grupos en las partes altas de la epidermis, junto con un infiltrado perivascular superficial (a veces también intersticial) linfohistiocitario, que puede acompañarse de neutrófilos y eosinófilos10 (fig. 2C,D). Esta histopatología es la que se evidencia también cuando las placas persistentes presentan un patrón clínico reticulado13.
Los queratinocitos necróticos han sido presentados en la literatura como fuente de error diagnóstico con un eritema multiforme17. Sin embargo, el eritema multiforme es una dermatitis de interfase en la que se espera al menos algo de vacuolización de la basal y en donde los queratinocitos necróticos se ven preferentemente en el estrato basal, independientemente de que algunos estén localizados en estratos superiores. Por el contrario, su localización es en estratos superiores de la epidermis en el Still. A pesar de eso, se ha de recordar que algunos raros casos de enfermedad de Still pueden mostrar un cuadro histopatológico de degeneración vacuolar de la capa basal con pústulas subcórneas e intracórneas18.
Algunos casos con presentación clínica de pápulas y placas parduscas persistentes han mostrado en la biopsia depósito de mucina en la dermis reticular19, un hallazgo también publicado en casos de rash maculopapular lineal20.
Los cuadros clínicamente urticariformes presentan las características histopatológicas típicas de la urticaria, con edema dérmico e infiltrado perivascular (a veces también intersticial) rico en neutrófilos, sin vasculitis14.
Otro patrón histopatológico visto en los casos atípicos (principalmente en los que cursan con eritema pruriginoso persistente diseminado) es un edema de la dermis media y alta con un infiltrado perivascular de predominio mononuclear en el que se observan algunos neutrófilos.
La descripción clínica en la literatura de prurigo pigmentosa-like se corresponde en la biopsia con áreas de paraqueratosis y espongiosis eosinofílica en una epidermis acantótica, células disqueratóticas aisladas en la epidermis y un infiltrado dérmico perivascular superficial constituido por neutrófilos, eosinófilos y linfocitos16.
Algunos casos atípicos pueden presentar espongiosis con edema dérmico acompañando al infiltrado perivascular dérmico. Estos cuadros suelen presentarse clínicamente como erupciones vesiculopustulosas.
Por último, en la literatura se han presentado algunos casos con afectación oral, con máculas marronáceas en la mucosa oral interna de los labios y en la lengua21. En ellos, la biopsia muestra un edema coriónico con un infiltrado perivascular rico en neutrófilos.
Concepto de enfermedad relacionada con la IgG4La enfermedad relacionada con la IgG4 (ERIgG4) es una entidad de relativamente reciente descripción que cursa con manifestaciones cutáneas y sistémicas22. De hecho, puede afectar a prácticamente cualquier órgano, siendo los más frecuentes el páncreas, las glándulas salivales y las lacrimales.
El marcador histopatológico de la ERIgG4 es un infiltrado linfoplasmocitario rico en células plasmáticas IgG4+ junto con una fibrosis de patrón estoriforme23 (fig. 3A,B). En concreto, la relación IgG4/IgG debe ser superior al 40% y deben contabilizarse más de 10 células plasmáticas IgG4+ por campo de gran aumento24. La fibrosis, por el contrario, no suele aparecer hasta que la enfermedad está avanzada y, por lo tanto, su ausencia no excluye el diagnóstico. La otra característica histopatológica frecuente en el cuadro es la flebitis obliterante.
A bajo aumento, la enfermedad IgG4-relacionada debe sospecharse por la coexistencia de un infiltrado rico en células plasmáticas con áreas de fibrosis concéntrica (H&E ×40). B)Infiltrado rico en células plasmáticas en un caso de enfermedad IgG4-relacionada (H&E ×400).")
Sin embargo, este marcador histopatológico (la riqueza de células plasmáticas IgG4+) es controvertido por sí solo fuera de un contexto clínico. Ello es así porque hay diversas entidades que pueden presentarse con riqueza de células IgG4+. Se ha sugerido que algunas de las entidades cutáneas ricas en células plasmáticas podrían ser en realidad formas de enfermedad relacionada con la IgG4; tal es el caso de la enfermedad de Rosai-Dorfman o de la hiperplasia angiolinfoide con eosinofilia23. Pero no todos los infiltrados ricos en IgG4 serían enfermedades IgG4-relacionadas: abundantes células plasmáticas IgG4 pueden verse en la inflamación crónica de la mucosa oral o en la artritis reumatoide25.
Hay evidencias de que las moléculas de IgG4 no son inflamatorias. Por lo tanto, más bien parecen la respuesta secundaria a un proceso primario (que podría ser la fibrosis) muy probablemente en individuos con predisposición genética26-29. Sin embargo, también podrían ser un fenómeno secundario a algunas enfermedades inflamatorias inmunes o a que los procesos inflamatorios crónicos pueden cursar con infiltrados ricos en células plasmáticas IgG4+, sin por ello necesariamente cumplir criterios de enfermedad IgG4-relacionada30.
Desde el punto de vista clínico, la enfermedad se presenta con engrosamientos, masas o tumores, localizados o difusos, en múltiples órganos31. En la piel la presentación es variada en forma de máculas, pápulas, placas, nódulos, púrpura, ampollas o rash, siendo la localización más frecuente la cabeza y el cuello32. Los dermatólogos han de estar alerta sobre este diagnóstico cuando estas manifestaciones asocien pancreatitis idiopática, fibrosis retroperitoneal, aortitis o ERIgG4 sistémica ya diagnosticada32.
Uno de los diagnósticos diferenciales más complicados es la plasmocitosis sistémica, una entidad más frecuente en Japón, que afecta varios órganos incluida la piel, en forma de erupción de pápulas rojizo-marronáceas de distribución preferentemente en tronco33. En la biopsia, la plasmocitosis sistémica se caracteriza por un infiltrado superficial y profundo, perivascular y perianexial de células plasmáticas maduras de expresión politípica de inmunoglobulinas. Dado que a veces la subpoblación IgG4 es prominente en este infiltrado, se ha sugerido si al menos algunos de estos casos podrían ser formas de enfermedad IgG4-relacionada34-36.
Por último, decir que la enfermedad IgG4-relacionada es una entidad de descripción reciente y por lo tanto no completamente comprendida en sus causas y en su patogenia y que muy probablemente comprende un grupo heterogéneo de condiciones que deberán ser caracterizadas en un futuro próximo37.
Nuevos avances en biopsia de lupusDesde el punto de vista clínico, el lupus eritematoso con afectación cutánea puede dividirse en dos grandes grupos: afectación exclusivamente cutánea (en la que se distinguen fundamentalmente el lupus eritematoso discoide y la forma subaguda) y el lupus sistémico (por ejemplo, el agudo), que afecta también a órganos internos.
Se ha discutido durante mucho tiempo si existen signos histopatológicos sugestivos de lupus eritematoso sistémico. También si existen formas de lupus exclusivamente limitadas a la piel, sin repercusión sistémica. La evolución a formas sistémicas de pacientes con lupus eritematoso discoide crónico indica que no existe una línea nítida entre ambos grupos, cuya distinción, sin embargo, se mantiene debido al bajo riesgo de progresión sistémica de muchas de las formas cutáneas de lupus.
Una de las formas de lupus que se admitía como primaria cutánea sin repercusión sistémica es el lupus túmido, siendo considerado como una entidad separada38. Sin embargo, recientemente se ha presentado un caso de lupus eritematoso túmido convertido desde lupus discoide que apoya la intercambiabilidad de las distintas variantes de lupus entre sí39. Por el contrario, otros autores defendieron que el lupus debía ser enfocado como una enfermedad sistémica con variable repercusión en distintos órganos, uno de los cuales es la piel40.
Desde el punto de vista histopatológico, existen cambios interpretados como signos de cronicidad o de agudeza. Así, mientras el lupus sistémico suele mostrar poca o ninguna atrofia epidérmica (fig. 4A), muy poca degeneración vacuolar de la basal (fig. 4B), ausencia de engrosamiento de la membrana basal, mínimos tapones córneos, mínimo edema dérmico y mínimo infiltrado perianexial (fig. 4C), todos esos caracteres son prominentes en el lupus eritematoso discoide41 (fig. 4D).
. Existe muy poca degeneración vacuolar de la basal sin evidencia de engrosamiento de la membrana basal (B: H&E ×400). El infiltrado inflamatorio dérmico es discreto y puede contener neutrófilos (C: H&E ×200). D)En contraste, el lupus eritematoso discoide muestra dilataciones infundibulares, tapones córneos prominentes e infiltrado perianexial marcado (H&E ×20).")
Afectación cutánea en un caso de lupus con manifestaciones sistémicas. Ausencia de atrofia epidérmica (A: H&E ×100). Existe muy poca degeneración vacuolar de la basal sin evidencia de engrosamiento de la membrana basal (B: H&E ×400). El infiltrado inflamatorio dérmico es discreto y puede contener neutrófilos (C: H&E ×200). D)En contraste, el lupus eritematoso discoide muestra dilataciones infundibulares, tapones córneos prominentes e infiltrado perianexial marcado (H&E ×20).
Además, el conocimiento de los últimos años nos ha conducido a un escenario patogénico de lesión cutánea muy diferente en las formas sistémicas y localizadas. Estos dos escenarios serían, no obstante, dinámicos y podrían modificarse durante la vida del paciente según factores externos e internos.
Así, la migración de linfocitos T citotóxicos y de células natural killer desde la circulación a la piel es una característica de las formas exclusivamente cutáneas pero no de las sistémicas41. Así mismo, las células dendríticas plasmacitoides, que tan útiles han resultado como marcador diagnóstico de lupus, son comúnmente abundantes en las formas cutáneas de lupus pero no tanto en las biopsias de piel en el contexto de un lupus sistémico, en donde estas células se mantendrían preferentemente confinadas a la circulación. Lo contrario sucede con los linfocitosT reguladores, que aparecen muy disminuidos en las biopsias de piel de lupus sistémico, pero no tan disminuidos en las de lupus con afectación exclusivamente cutánea41.
Las células dendríticas plasmacitoides producen un ambiente de exceso de interferón alfa y beta, que no son fáciles de medir en piel en la práctica diaria. Sin embargo, myxovirus resistance protein 1 (MxA) parece un buen marcador alternativo indicativo de los niveles de interferón en piel y sangre periférica42.
Algunos de los signos histopatológicos de las biopsias de lupus eritematoso pueden resultar de utilidad pronóstica, no solo diagnóstica. Por ejemplo, existe una estrecha correlación entre el número de queratinocitos apoptóticos y la actividad del lupus eritematoso41 (fig. 5A). Hay mayor actividad apoptótica en el lupus subagudo y en el sistémico, dos de las formas que se asocian a mayor actividad sistémica43. Además, la acumulación de apoptosis parece ser crucial como detonante de la lesión cutánea41, contribuyendo a la inflamación y a la inmunidad al liberar moléculas proinflamatorias como interferón alfa, interleucina-1 y HMGB144.
Numerosas figuras apoptóticas en la basal epidérmica (H&E ×200). Se ha correlacionado el número de queratinocitos apoptóticos con la actividad lúpica. B)Despegamiento subepidérmico en un caso de lupus ampolloso (H&E ×40). C)Presencia de numerosos polimorfonucleares en el infiltrado crónico de un lupus ampolloso (H&E ×400). D)Infiltrado perivascular en una lesión ulcerada correspondiente a una afectación cutánea por enfermedad de Kikuchi (H&E ×40).")
A)Numerosas figuras apoptóticas en la basal epidérmica (H&E ×200). Se ha correlacionado el número de queratinocitos apoptóticos con la actividad lúpica. B)Despegamiento subepidérmico en un caso de lupus ampolloso (H&E ×40). C)Presencia de numerosos polimorfonucleares en el infiltrado crónico de un lupus ampolloso (H&E ×400). D)Infiltrado perivascular en una lesión ulcerada correspondiente a una afectación cutánea por enfermedad de Kikuchi (H&E ×40).
Una de las variantes infrecuentes de lupus es la ampollosa, de la que recientemente se ha estudiado una serie grande de pacientes, demostrando que debe ser considerada como afectación sistémica, asociada a nefritis lúpica en la mitad de los casos45. Desde el punto de vista histopatológico, además del despegamiento subepidérmico (fig. 5B) es especialmente interesante el infiltrado rico en polimorfonucleares neutrófilos45 (fig. 5C). No es sorprendente, por lo tanto, la buena respuesta obtenida con dapsona en el 90% de los casos45.
Por el contrario, hay entidades controvertidamente clasificadas como formas de lupus eritematoso. Una de ellas es el síndrome antifosfolípido, frecuentemente sobrediagnosticado como lupus eritematoso y, en consecuencia, tratado inadecuadamente. Para evitar esto, algunos autores han sugerido incluir en futuras clasificaciones el requerimiento de al menos un criterio de lupus eritematoso sistémico antes de realizar el diagnóstico de lupus en pacientes con síndrome antifosfolípido46.
Al igual que otras enfermedades de componente predominantemente autoinmune, el lupus eritematoso ha sido ocasionalmente asociado a entidades de supuesta etiopatogenia autoinflamatoria, tales como la hidradenitis supurada47.
Recientemente se ha sugerido la posibilidad de usar microscopia confocal fluorescente ex vivo en intraoperatoria para el diagnóstico rápido de enfermedades inflamatorias, incluido el lupus, con el fin de no diferir el tratamiento48.
Las manifestaciones cutáneas de lupus van acompañadas de un incremento de la carcinogénesis cutánea por diferentes motivos, incluyendo la presencia de cicatrización crónica, la inflamación persistente, la sensibilidad a las radiaciones ultravioletas o el uso de inmunosupresores para el manejo terapéutico49. Sin embargo, tanto observaciones clínicas de co-localización de múltiples cánceres cutáneos independientes en áreas de inflamación activa en pacientes con lupus eritematoso discoide seguidos durante años, así como experimentación en modelos murinos, conducen a la conclusión de que la carcinogénesis está estrechamente relacionada con la inflamación y de que la supresión o limitación de esta conlleva una disminución considerable del riesgo de desarrollo de tumores49.
Lupus y enfermedad de Kikuchi-FujimotoLa enfermedad de Kikuchi-Fujimoto (EKF) (linfadenitis necrosante histiocítica) es un trastorno idiopático. La hipótesis etiopatogénica más aceptada es la viral/autoinmune50, y está basada en hallazgos clínicos (curso autolimitado), de laboratorio (linfocitos atípicos en sangre periférica, elevación de citocinas, interferón alfa e interleucina-6)51 e histopatológicos (infiltrado preferentemente de linfocitosT-CD8 con apoptosis y un fondo necrótico)52-54. Se trataría o bien de una enfermedad autoinmune o bien de una respuesta inmune exagerada a una infección vírica. Aunque el proceso suele ser autorresolutivo, pueden recidivar incluso muchos años después del primer episodio55.
La EKF guarda una controvertida relación con el lupus eritematoso sistémico y en ocasiones ambas entidades han coexistido en el mismo paciente. En dichos casos la EKF puede ser previa, simultánea o posterior al lupus56. En otras ocasiones, un paciente diagnosticado de EKF cumple criterios de lupus en alguno de sus episodios57,58.
Adicionalmente, los cambios evidenciados en las adenopatías de pacientes con lupus sistémico y de pacientes con EKF son superponibles, y en muchas ocasiones no pueden ser distinguidos58. Uno de los hallazgos morfológicos que se ha defendido como más discriminatorio entre el lupus y la EKF es la presencia de cuerpos hematoxilínicos que remedan agregados de ADN y anticuerpos anti-ADN59. Los cuerpos hematoxilínicos han sido principalmente descritos en el ganglio linfático pero también pueden verse en biopsias de otros órganos, como la médula ósea o el músculo esquelético60,61. Sin embargo, no son una característica común de las biopsias cutáneas de lupus eritematoso.
Por el contrario, la biopsia cutánea en casos de EKF muestra un infiltrado linfocitario perivascular e intersticial (fig. 5D) con presencia de células histiocitoides CD163+, CD68+ que expresan mieloperoxidasa62 y cuya naturaleza está en la actualidad bajo investigación. Son característicos los focos de necrosis sin neutrófilos maduros, así como la lesión dermoepidérmica de interfase59,63. Esta última es un signo en común con el lupus, como lo son también el depósito de mucina o la paniculitis59.
Por todos estos solapamientos, se ha considerado la posibilidad de que la EKF sea en realidad una forma frustrada de lupus eritematoso64, con un curso autolimitado y autorresolutivo en un período de varias semanas o varios meses en la mayor parte de los casos55,65. Quedarían, pues, una minoría de casos de mala evolución a formas severas sistémicas de EKF, algunas de ellas ocasionalmente fatales, con respuesta solo a tratamiento precoz e intenso de terapia inmunosupresora, así como los casos de «solapamiento con» o «evolución a» lupus eritematoso sistémico.
Biopsia cutánea en enfermedades digestivasProbablemente la enfermedad digestiva de repercusión cutánea más conocida sea la intolerancia al gluten, con la dermatitis herpetiforme como reflejo asociado. La presencia de IgA granular en la dermis papilar es debida a inmunocomplejos entre IgA con avidez por la piel y la enzima transglutaminasa66.
En 2006, Humbert et al.67 propusieron su clasificación de enfermedades cutáneas asociadas a la intolerancia al gluten en cuatro grandes grupos según su mecanismo patogénico. Entre las de mecanismo alérgico están la dermatitis atópica68 y la urticaria69 (aguda o crónica). Entre las autoinmunes, la psoriasis70-73. Otras entidades asociadas son la rosácea y la estomatitis aftosa74,75. Aunque la lista de asociaciones es bastante más larga, muchas entidades han sido asociadas solo de modo esporádico o fortuito.
Otro gran grupo de condiciones digestivas con repercusión cutánea son las enfermedades inflamatorias del intestino grueso, con manifestaciones cutáneas de tipo granulomatoso o reactivo, pero también en ocasiones asociadas a deficiencias nutricionales o a iatrogenia76. Excluyendo las dos últimas (nutricional e iatrogénica), se observa afectación cutánea en el 14,9% de los pacientes con enfermedad inflamatoria intestinal (Crohn o colitis ulcerosa)76.
Se considera como específica la afectación de las áreas de la piel más inmediatas al tracto gastrointestinal por granulomas no caseificantes de la enfermedad de Crohn, a veces acompañándose de fístulas o abscesos77 (fig. 6A). Estas mismas manifestaciones granulomatosas pueden verse, no obstante, alejadas de los orificios anal y bucal, dando lugar al llamado Crohn metastásico. Este último puede aparecer incluso en pacientes con las manifestaciones digestivas bien controladas con tratamiento78.
Trayecto fistuloso en la biopsia de una lesión de Crohn «metastásico» en pierna (H&E ×20). B)Colagenosis perforante con eliminación de fibras eosinofílicas de colágeno por un cráter de material necrótico (H&E ×20). C)Calcifilaxis mostrando las paredes calcificadas de vasos subcutáneos de calibre pequeño (H&E ×200).")
A)Trayecto fistuloso en la biopsia de una lesión de Crohn «metastásico» en pierna (H&E ×20). B)Colagenosis perforante con eliminación de fibras eosinofílicas de colágeno por un cráter de material necrótico (H&E ×20). C)Calcifilaxis mostrando las paredes calcificadas de vasos subcutáneos de calibre pequeño (H&E ×200).
Por el contrario, se consideran reactivas las afectaciones que no muestran las características granulomatosas típicas del Crohn pero que son probablemente la manifestación cutánea del trastorno autoinflamatorio subyacente. Muchas presentan, por lo tanto, un importante infiltrado inflamatorio dérmico de polimorfonucleares neutrófilos, como el pioderma gangrenoso, el síndrome de Sweet, el síndrome de dermatosis-artritis asociado al intestino, la piodermatitis-pioestomatitis vegetans, el síndrome SAPHO (sinovitis, acné, pustulosis, hiperostosis, osteítis) y el síndrome PAPA (artritis piogénica, pioderma gangrenoso, acné)77. Se incluye también como reactiva el eritema nodoso, de histopatología característica con su paniculitis preferentemente septal.
Las formas secundarias a déficits nutricionales se consideran sobre todo debidas a carencias de vitaminasA, B12 y C79. Por último, los fármacos usados en el tratamiento de la enfermedad inflamatoria intestinal son responsables de manifestaciones cutáneas diversas.
Las enfermedades autoinmunes hepáticas, tales como la cirrosis biliar primaria, la colangitis esclerosante primaria o la hepatitis autoinmune, muestran asociación con diversas manifestaciones cutáneas: la relación con el vitíligo está demostrada80, y otras asociaciones probables son la alopecia areata, la psoriasis, el pioderma gangrenoso o la amiloidosis81. Contrariamente a como se pensó, la colangitis biliar primaria no está relacionada con el liquen plano pero sí con la pustulosis amicrobiana de los pliegues80.
Recientemente se ha descrito que el virusE de la hepatitis muestra un tropismo endotelial capaz de conducir a los linfocitosT a la clonalidad y desencadenar distintos tipos de linfoma82.
Por último, recordar que los tratamientos antivirales en la hepatitis son capaces de desencadenar enfermedades sistémicas como el lupus eritematoso83 o la crioglobulinemia84.
Biopsia cutánea en enfermedades renalesLas manifestaciones cutáneas asociadas a enfermedad renal pueden ser clasificadas en tres categorías: la enfermedad renal terminal, las manifestaciones de la uremia y las asociadas al trasplante. Entre el 50-100% de los pacientes con enfermedad renal terminal presentan algún tipo de manifestación cutánea.
Algunas de las manifestaciones dermatológicas están directamente relacionadas con la enfermedad primaria de base (tabla 1), cual es el caso de la diabetes, el lupus eritematoso, la púrpura de Henoch-Schönlein, la granulomatosis con poliangeítis, la poliarteritis nodosa, la endocarditis bacteriana, la amiloidosis85, las hepatitis víricas o la esclerosis sistémica, entre otras.
Enfermedades sistémicas con afectación renal y cutánea
| Trastorno sistémico | Afectaciones cutáneas |
|---|---|
| Diabetes mellitus | Infecciones cutáneasNecrobiosis lipoídicaDermopatía diabéticaBullosis diabeticorumSíndrome de engrosamiento cutáneoAcantosis nigricansXantomas eruptivosEnfermedad de KyrleManifestaciones de la vasculopatía diabéticaManifestaciones de la neuropatía diabética |
| Lupus eritematoso | Forma crónicaForma subagudaForma agudaFormas ampollosasForma túmidaÚlceras mucosasPúrpuraUrticariaAtrofia blancaDegosLivedo reticularisTromboflebitisRaynaudCutis laxaAnetodermiaMucinosis papularPustulosis amicrobiana de las flexurasPerniosisDermatosis neutrofílica |
| Henoch-Schönlein | PúrpuraExantema maculopapuloso o urticariformeFormas ampollosas |
| Granulomatosis con poliangeítis | Nódulos subcutáneosÚlcerasIsquemia digitalVasculitisPaniculitis |
| Poliarteritis nodosa | Livedo reticularisNódulos subcutáneos dolorososÚlcerasPúrpuraNecrosis cutáneaAutoamputaciones |
| Endocarditis bacteriana subaguda | Lesiones palmoplantares de JaanewayNódulos subcutáneos digitales de OslerManchas retinianas de RothPetequias conjuntivales y en mucosasHemorragias ungueales en astilla |
| Amiloidosis | Púrpura periorbitaria y en grandes plieguesPápulas blancasLesiones esclerodermiformesDistrofia unguealAlopeciaLesiones ampollosasMacroglosia |
| Hepatitis víricas | PúrpuraIctericiaEritema palmarLiquen planoVasculitis urticarialCrioglobulinemiaEritema nodosoEritema multiformeSíndrome de BehçetPorfiria cutánea tardaHipertricosisPlacas esclerodermiformesGranuloma anular generalizadoPoroqueratosisEritema acral necrolíticoPoliarteritis nodosa |
| Esclerosis sistémica | EsclerodermiaEdema crónicoEsclerodactiliaCicatrices de pulpejosRaynaudTelangiectasiasHipopigmentación e hiperpigmentación cutáneaAlopeciaAnhidrosis |
Entre los efectos asociados a la uremia están la xerosis, el prurito, la púrpura, la pigmentación cutánea, la calcifilaxis, las enfermedades perforantes, la porfiria y la pseudoporfiria, las esclerosis relacionadas con la acumulación de gadolinio y la alopecia. También pueden observarse distintas alteraciones ungueales, como las uñas mitad-mitad (uñas de Lindsay), las líneas de Muehrcke, la ausencia de lúnula, la palidez ungueal o los trastornos en la coloración de la lámina86.
Por último, las principales afecciones cutáneas relacionadas con el trasplante renal, además del rechazo, son los trastornos asociados con la medicación (cambios cushingoides, hiperplasia gingival, cambios en la unidad pilosebácea…) y los relacionados directamente con la inmunosupresión (infecciones de distintas etiologías, tumores cutáneos, poroqueratosis…).
Uno de los trastornos renales con una traducción cutánea asociada es el síndrome de Reed, con tendencia a padecer carcinoma renal papilar. Los pacientes con el síndrome asocian leiomiomas cutáneos en los que se puede demostrar la inmunotinción para la fumarato hidratasa87.
La insuficiencia renal crónica guarda una estrecha asociación con las enfermedades cutáneas perforantes, caracterizadas por la eliminación transepidérmica de distintos componentes dérmicos (fibras elásticas, de colágeno…). Tradicionalmente se han clasificado en cuatro grandes grupos: elastosis perforans serpiginosa, colagenosis perforante reactiva, foliculitis perforante y enfermedad de Kyrle, siendo las cuatro englobadas por varios autores bajo el epígrafe global de «dermatosis perforante adquirida».
La patogenia de estas dermatosis perforantes es desconocida aunque probablemente el rascado tiene mucho que ver en un número significativo de pacientes, como comprueba el hecho de que tras tratamiento para mitigar el prurito las lesiones suelen disminuir mucho en número.
Desde el punto de vista de la histopatología, las dermatosis perforantes se presentan como invaginaciones verticales de la epidermis en forma de un canal corto con epidermis acantótica a ambos lados. La epidermis de la base del canal puede aparecer preservada o en ocasiones erosionada. En el canal hay material degenerado basofílico con neutrófilos y fibras de tejido conectivo (colágeno en las colagenosis, elásticas en las elastosis, ninguna en la enfermedad de Kyrle… e incluso fibras elásticas y colágenas coexistiendo) (fig. 6B). En el caso de la elastosis perforans serpiginosa inducida por D-penicilamina las fibras elásticas adoptan un patrón arrosariado característico (lumpy-bumpy).
Otro de los cuadros histopatológicos cruciales en el contexto de la enfermedad renal es la calcifilaxis, una enfermedad con alta tasa de mortalidad. Aunque de patogénesis desconocida, la calcificación de paredes vasculares de capilares y vasos de pequeño-mediano calibre, generalmente subcutáneos (fig. 6C), parece ser la principal responsable del flujo crónico que provoca necrosis aguda isquémica dérmica y subcutánea, acompañándose de trombosis vasculares que probablemente traducen un estado de hipercoagulabilidad. Como resultado, la angioplasia dérmica es un hallazgo histopatológico frecuentemente acompañante.
La demostración de la calcifilaxis en la biopsia requiere un punch profundo que permita evaluar los vasos de la hipodermis. Sin embargo, en ocasiones se observa un hallazgo histopatológico dérmico más superficial patognomónico de la calcifilaxis: las calcificaciones periecrinas «en polvo». Adicionalmente, las calcificaciones neurales pueden también verse.
Por último, remarcar que la presencia calcifilaxis no es sinónimo de enfermedad renal: los hallazgos histopatológicos cutáneos de las calcifilaxis evidenciados en pacientes con y sin enfermedad renal parecen ser similares.
Biopsia cutánea en sarcoidosis: el concepto de granuloma por síliceUno de los aspectos más controvertidos en la literatura es el hallazgo de granulomas sarcoideos en una biopsia cutánea, y si guarda alguna relación con una sarcoidosis sistémica.
Muchos de estos granulomas, cuando son examinados con luz polarizada, contienen partículas birrefringentes de sílice (fig. 7A,B). Esto ha dado lugar al concepto de granuloma por sílice88. Muy recientemente se han caracterizado físico-químicamente estos depósitos de los granulomas sarcoideos, siendo identificados como sílice cristalina en áreas centrales del granuloma y como calcita en su periferia89.
Reacción granulomatosa epitelioide sarcoidea en la dermis en un caso de granuloma por sílice (H&E ×100). B)En el examen de los granulomas sarcoideos con luz polarizada se observan numerosas partículas intensamente birrefringentes (H&E ×100, luz polarizada). C)Ejemplo típico de un tricolemoma (H&E ×20); incluso a este aumento se pueden ver las vacuolizaciones citoplásmicas celulares características. D)Preservación de la expresión de PTEN en un tricolemoma esporádico, no asociado a síndrome de Cowden (PTEN ×20).")
A)Reacción granulomatosa epitelioide sarcoidea en la dermis en un caso de granuloma por sílice (H&E ×100). B)En el examen de los granulomas sarcoideos con luz polarizada se observan numerosas partículas intensamente birrefringentes (H&E ×100, luz polarizada). C)Ejemplo típico de un tricolemoma (H&E ×20); incluso a este aumento se pueden ver las vacuolizaciones citoplásmicas celulares características. D)Preservación de la expresión de PTEN en un tricolemoma esporádico, no asociado a síndrome de Cowden (PTEN ×20).
Para algunos autores, la mera presencia de sílice excluye una sarcoidosis sistémica90-92. Por el contrario, otros estudios han demostrado que las partículas de sílice son frecuentes en los granulomas cutáneos de pacientes con sarcoidosis sistémica93,94.
La sílice es un material prácticamente ubicuo en la naturaleza. Muy probablemente penetra en nuestra piel muy temprano en nuestras vidas, fruto de traumatismos leves durante la infancia, con un largo periodo de latencia95. Tan es así, que lo sorprendente es que las reacciones granulomatosas al sílice no sean más comunes.
Algunos estudios han demostrado que las partículas de sílice son tan frecuentes en los granulomas cutáneos sarcoideos asociados a sarcoidosis como en los no asociados96. Además, cuando se estudian biopsias de piel con diagnósticos dispares, sin relación con la sarcoidosis ni con la inflamación granulomatosa, se demuestran partículas de sílice en casi el 40% de ellas96.
En consecuencia, se ha llegado a sugerir que aunque las partículas de sílice serían un detonante, hace falta una predisposición, posible en los pacientes con sarcoidosis y, por lo tanto, más susceptibles de desarrollar granulomas ante ciertos estímulos97. Este mecanismo ha sido corroborado por la descripción de procesos granulomatosos similares en pacientes con sarcoidosis frente a otro tipo de partículas, como por ejemplo tatuajes98, tratamientos de mesoterapia99, catéteres100 o, más recientemente, por el desarrollo de granulomas sarcoideos, en pacientes predispuestos, a nivel de las zonas de venopunción101. Un proceso similar se ha descrito en otros órganos frente a otro tipo de partículas (como talco) en pacientes con tendencia a otro tipo de granulomatosis, como por ejemplo la enfermedad de Crohn102.
Tricoepiteliomas múltiples y síndrome de CowdenEl síndrome de Cowden es una genodermatosis de herencia autosómica dominante y afectación multiorgánica que entraña un riesgo de desarrollo de neoplasias, sobre todo de mama, tiroides y endometrio103. La expresividad clínica del síndrome es variable, al igual que las mutaciones identificadas hasta la fecha104,105. El nombre lo recibió no de los clínicos que describieron la entidad, sino de la paciente en la que fue descrita —Rachel Cowden—, quien murió después de un cáncer de mama106. Además, el síndrome se caracteriza por hamartomas múltiples de muchas localizaciones, como mucosa oral, piel, tracto gastrointestinal, hueso, ojos, sistema nervioso central y tracto urinario107.
El marcador cutáneo del síndrome de Cowden son los tricolemomas múltiples faciales (más de tres) (fig. 7C), considerados marcador patognomónico del síndrome. Otros marcadores patognomónicos son la queratosis acral, la papilomatosis oral o la enfermedad de Lhermitte-Duclos.
Aunque la etiología es desconocida, hasta en el 85% de los casos se observa una mutación del gen supresor tumoral del homólogo del fosfato y la tensina (PTEN). Su producto, la proteína PTEN, desempeña funciones no del todo conocidas, modulando el ciclo celular y la supervivencia celular. Su inactivación conduce a un crecimiento y a una supervivencia celulares excesivos.
Se ha sugerido que el PTEN podría ser usado como marcador inmunohistoquímico de síndrome de Cowden, ya que hay pérdida de su expresión en hasta el 83% de casos sindrómicos108 (fig. 7D). Sin embargo, su pérdida no es patognomónica, ya que puede observarse en entre el 3 y el 13% de casos esporádicos108,109. Adicionalmente, existen formas de Cowden que no están asociadas a la mutación de PTEN110.
Recientemente se ha descrito una presentación segmentaria de colagenomas estoriformes como parte del espectro sindrómico111.
Actitud ante un posible síndrome de Muir-TorreEl síndrome de Lynch —también conocido como «cáncer colorrectal hereditario sin poliposis»— es una condición autosómica dominante asociada con una mutación de línea germinal en los genes reparadores del ADN, responsable de entre el 5 y el 10% de los cánceres colorrectales. El 90% de los cánceres colorrectales que se desarrollan en pacientes con síndrome de Lynch están relacionados con el mecanismo patogénico de la inestabilidad de microsatélites (IMS). Los microsatélites son secuencias repetidas de 1 a 6 pares de bases de longitud que aparecen normalmente en nuestro ADN. Lo interesante es que aunque varían de longitud de una persona a otra, su longitud es constante en una persona concreta. Si hay defectos en las proteínas reparadoras de ADN, encontraremos variación de longitud en los microsatélites de una persona. Esa condición no es directamente causante de un riesgo tumoral sino más bien un resultado, un marcador y una alerta del mal funcionamiento del sistema de reparación del ADN.
Los pacientes con síndrome de Lynch tienen un alelo funcional para la reparación del ADN y un alelo defectuoso. El síndrome se desarrolla tras la mutación somática incapacitante del alelo funcional112. Como dicha vía implica una reparación deficiente de errores genéticos en general, los individuos con síndrome de Lynch tienen riesgo de padecer otros cánceres a una edad temprana: endometrial el más frecuente, y en menor escala de ovario, estómago, intestino delgado, páncreas, tracto hepatobiliar, tracto urinario, próstata, cerebro y piel.
La presentación de tumores cutáneos asociados a IMS se conoce como síndrome de Muir-Torre (SMT), por lo que este último puede considerarse una variante del síndrome de Lynch. El SMT tiene un tipo de herencia autosómica dominante en el 59% de los casos, con un alto grado de penetración y expresión variable. Los tumores cutáneos asociados más frecuentes son los sebáceos y los queratoacantomas. Entre los primeros, el adenoma sebáceo, el sebaceoma, el carcinoma sebáceo y el carcinoma basocelular con diferenciación sebácea. Se excluye el nevus sebáceo de Jadassohn. Aunque la hiperplasia sebácea guarda relación con el SMT, suele dejarse fuera de esta lista por lo frecuente que es en pacientes sin el síndrome. Algo similar sucede con el queratoacantoma (tanto solitario como múltiple), el carcinoma espinocelular y los quistes infundibulares múltiples, todos ellos asociados a SMT pero tan frecuentes en la población general que su carácter como marcador está por ello mermado. No obstante, algunos autores sugieren investigar un posible SMT en los casos de queratoacantoma múltiple o de queratoacantoma con diferenciación sebácea. Recientemente se ha descrito un subgrupo de tumores sebáceos con un patrón morfológico peculiar («carcinoide-like») que parece no guardar relación con el SMT113.
El adenoma sebáceo es la neoplasia sebácea más frecuentemente asociada a SMT. Se trata de un tumor multilobulado dérmico. Cada lóbulo aparece rodeado por una pseudocápsula de tejido conectivo y está constituido mayoritariamente por células multivacuoladas de aspecto sebáceo maduro (fig. 8B). En la periferia de cada lóbulo se observan varias capas de células basófilas germinativas (en contraste con la hiperplasia sebácea que, como mucho, muestra dos capas de células germinativas; fig. 8A). Estas últimas pueden mostrar disposición en empalizada en algunas ocasiones, aunque no es lo habitual. A diferencia del adenoma sebáceo, el sebaceoma está mayoritariamente constituido por células basaloides maduras y no suele presentar una arquitectura lobular (fig. 8C). Sin embargo, existe un continuo morfológico entre adenoma sebáceo y sebaceoma, por lo que algunos autores utilizan el término unificador de sebomatricoma. Lo importante es que ambas neoplasias muestran una arquitectura general simétrica (contrario al carcinoma sebáceo) y que desde el punto de vista citológico no muestran pleomorfismo nuclear (fig. 8D). Del mismo modo, la necrosis es un importante indicador de malignidad en las neoplasias sebáceas, todo lo contrario que las mitosis, que pueden verse en abundancia incluso en tumores sebáceos benignos.
Hiperplasia sebácea (H&E ×20). Neoplasias sebáceas asociadas al SMT: B)adenoma sebáceo (H&E ×40); C)sebaceoma (H&E ×40), y D)carcinoma sebáceo (H&E ×200).")
Las tres neoplasias sebáceas mencionadas arriba deben diferenciarse del carcinoma basocelular con diferenciación sebácea, un tumor que muestra muchos de los atributos del basocelular clásico (empalizada periférica, retracción estromal, estroma con mucina…). La diferenciación sebácea suele verse en forma de sebocitos maduros en variable cantidad.
Se debe aclarar que los tumores sebáceos asociados al SMT muestran no infrecuentemente características morfológicas ambiguas que no permiten su clasificación nítida como benignos o malignos. Esta cuestión no suele ser, no obstante, crítica en tanto en cuanto la neoplasia haya sido extirpada en su totalidad. El SMT es un ejemplo de IMS114,115.
Para muestrear esta condición de riesgo se podrían secuenciar los microsatélites del individuo, un proceso costoso y laborioso para ser usado como cribado. Existen por lo tanto alternativas más prácticas. En concreto, la determinación de la expresión de proteínas reparadoras del ADN. El National Cancer Institute recomienda la inclusión de cinco marcadores para la detección de individuos con IMS116: el MutL Homolog1 (MLH1), el MutS protein homolog2 (MSH2), el MutS homolog6 (MSH6), el Postmeiotic Segregation Increased2 (PMS2) y el MutL Homolog3 (MLH3). Los genes que codifican para estas proteínas pertenecen al sistema de reparación del ADN (del inglés mismatch repair [MMR]). Se trata de un sistema de gran importancia evolutiva, por lo que aparece muy preservado desde las bacterias hasta los humanos.
El 90% de los casos de síndrome de Lynch están asociados a defectos del MLH1 y MSH2, el 7-10% al MSH6 y menos del 5% al PMS2117. La asociación con MLH3 es muy pequeña (menos del 3%), por lo que este último marcador no suele ser incluido en el estudio por la mayor parte de laboratorios. Alternativamente, el síndrome de Lynch puede ser el resultado de alteraciones epigenéticas112, así como de mutaciones somáticas esporádicas en ambos alelos (raro)118.
Es interesante que la mayor parte de pacientes con tumores cerebrales muestran mutaciones en MSH2 con preservación de MLH1 y presentan una historia familiar de tumores cerebrales119,120, por lo que tal posibilidad debería investigarse en el interrogatorio clínico ante dicho fenotipo.
En el momento actual, la mayor parte de hospitales realiza un primer estudio sistemático de cribado en todos los carcinomas colorrectales, endometriales y de intestino delgado mediante inmunohistoquímica para MSH2, MSH6, MLH1 y PMS2 (fig. 9). También sobre adenomas sebáceos, carcinomas sebáceos, sebaceomas y basocelulares con diferenciación sebácea. Es todavía motivo de discusión si deberían incluirse los queratoacantomas o las hiperplasias sebáceas.
 y MLH1 (B: ×20) con pérdida de la expresión de MSH2 (C: ×20) y MSH6 (D: ×20).")
Independientemente de dicho cribado, los pacientes con algún tumor de los mencionados deben ser interrogados para ver si cumplen criterios de Bethesda o de Amsterdam o, alternativamente, evaluar el score de riesgo de la Clínica Mayo para SMT121. En caso afirmativo, pueden estudiarse en un primer paso los marcadores de reparación del ADN mediante inmunohistoquímica, y si los resultados fuesen no concluyentes, realizar análisis genéticos y moleculares para descartar IMS.
La inactividad de alguno(s) de los genes que codifican para proteínas reparadoras del ADN puede también ser debida a hipermetilación de su promotor. En ese caso, la búsqueda de una mutación será fútil. Dado que las mutaciones de BRAF son frecuentes en los cánceres colorrectales esporádicos pero infrecuentes en los asociados al síndrome de Lynch, se ha sostenido que su estudio sería un paso útil previo al de los marcadores de reparación del ADN122. Sin embargo, un estudio sobre la mutación V600E en BRAF (la más frecuentemente investigada en los laboratorios) demostró BRAF «wild-type» en todas las neoplasias sebáceas asociadas a SMT investigadas123.
También se ha sugerido el uso de BRAF posterior a la inmunohistoquímica para proteínas reparadoras en los casos con pérdida de expresión de MLH1 no asociada a MSH2 por la alta asociación existente entre la mutación BRAF V600E y la metilación del promotor de MLH1124.
La pregunta clave es si se debe realizar un estudio sistemático de todos los tumores sebáceos extirpados o solo de los asociados a SMT, algo que se discute a fecha actual en la literatura. Lo más aceptado es que solo se debe incluir en el estudio el adenoma sebáceo, el sebaceoma y el carcinoma sebáceo. Como hemos mencionado, gran parte de los motivos de dejar fuera del estudio a hiperplasias sebáceas o a queratoacantomas obedece a su alta frecuencia de presentación en la población sin el síndrome. Sin embargo, el estudio inmunohistoquímico con los cuatro marcadores de reparación del ADN es en estos momentos una prueba económicamente muy asequible, por lo que parece justificado incluir al menos a las hiperplasias sebáceas en estos estudios.
Concepto de BAPomaBAP-1 es la proteína asociada al gen supresor de tumores BCRA-1 (BreastCancer-1). Este último codifica para la proteína reparadora de daño del ADN BCRA-1. BAP-1 se une a BRCA-1 para formar un complejo reparador de la función antitumoral125.
Los defectos en el gen BAP-1 pueden presentarse como condición familiar heredada de modo autosómico dominante con penetrancia todavía no determinada, en el que los individuos afectos desarrollan varios tipos de tumores tras la mutación somática del alelo funcional126, mostrando predisposición a melanomas uveales y cutáneos pero también a mesotelioma, colangiocarcinoma, carcinoma renal y carcinomas basocelulares.
Los nevi BAP-1-negativos fueron descritos como marcador cutáneo del síndrome. Se presentan como lesiones múltiples (entre 5 y 50), papulares, generalmente en la segunda década126. Lo interesante desde el punto de vista anatomopatológico es que pueden ser identificados por sus características morfológicas (aunque antes de conocerse la entidad, probablemente la mayoría —cuando no todos— fueron catalogados como nevi de Spitz). Se trata de nevi preferentemente dérmicos con amplio citoplasma y nucléolo prominente127. Con el estudio inmunohistoquímico para BAP-1 se observa pérdida de la positividad nuclear: de ahí lo paradójico de su designación como «BAPomas», cuando en realidad no expresan BAP. Una alternativa a este término es el epónimo de nevus de Wiesner, el primer autor que lo describió126,128.
Tampoco es infrecuente la presentación como nevus combinado en el que una parte negativa para BAP-1 coexiste con una parte de morfología no spitzoide convencional BAP-1-positiva129.
Al contrario que los nevi de Spitz, los nevi de Wiesner presentan mutaciones BRAF130,131. Sin embargo, como casi todo en dermatopatología, toda la información debe contextualizarse, ya que no todos los tumores melanocíticos que muestran pérdida de BAP-1 son nevi de Wiesner: se han descrito nevi de Spitz convencionales con pérdida de expresión de BAP-1129.
Como muchos otros marcadores, los nevi de Wiesner no siempre se presentan en el contexto de enfermedad familiar, sino que pueden mostrar una presentación esporádica.
Recientemente se ha publicado un estudio multicéntrico con las características clínicas y dermatoscópicas comunes a 48 nevi de Wiesner de 31 pacientes, concluyendo que se debería sospechar la entidad ante pápulas cupuliformes con áreas amorfas rosado-parduscas y periferia que muestren o bien glóbulos/puntos irregulares o un entramado también irregular132.
Desde el punto de vista histopatológico los nevi de Wiesner muestran simetría a bajo aumento, aunque a medida que los observamos más de cerca es evidente que están constituidos por diversos tipos de poblaciones celulares con predominio de melanocitos epitelioides de citoplasma amplio y núcleo atípico hipercromático. No se objetiva «maduración» melanocítica, es decir, los melanocitos de zonas más profundas no son más pequeños y monótonos que los de las partes altas del nevus. Frecuentemente incluso puede darse la paradoja contraria: parece haber un fenómeno de maduración inversa, con poblaciones de células más pequeñas en la parte alta, reflejando cierta estructuración caótica que presentan estos nevus. La población melanocítica puede acompañarse de un moderado infiltrado inflamatorio.
ConclusionesLas manifestaciones cutáneas precoces de enfermedades sistémicas permiten un diagnóstico temprano de estas entidades, con el consiguiente abordaje terapéutico adecuado. Un enfoque distinto en los últimos años de algunas de ellas nos ha permitido su mejor abordaje, catalogación y comprensión.
Conflicto de interesesEl autor declara no tener ningún conflicto de intereses.


