




INTRODUCCION
En 1953, Costa1 describió, bajo el término acroqueratolelastoidosis (AQE), la aparición, tanto esporádica como familiar, preferentemente en adolescentes o adultos jóvenes, de pápulas queratósicas de superficie umbilicada y traslúcida en el borde de la unión dorsopalmar de manos, y acentuación de los pliegues cutáneos en nudillos. Histológicamente las lesiones mostraban hiperqueratosis e hipergranulosis focal sobre un área deprimida de epidermis, asociadas a degeneración y fragmentación de las fibras elásticas subyacentes2.
En 1983, Dowd et al3 revisaron los casos publicados de AQE y describieron en 15 de ellos un subtipo al que denominaron hiperqueratosis focal acral (HFA) para diferenciarlos de la AQE original. Observaron en todos los pacientes las típicas lesiones papulosas, ovales o poligonales, crateriformes, a lo largo de los bordes de manos y pies. En dos de ellos existía afectación de los pliegues de las muñecas. Histológicamente se apreciaba hiperqueratosis focalizada sobre una depresión crateriforme en una epidermis ligeramente acantótica, pero no presentaba la elastorrexis típica que existe en la AQE, ni alteraciones del colágeno4-6. De los 15 pacientes descritos, 14 eran de raza negra y uno árabe; 12 eran mujeres y 3 hombres, con un rango de edades comprendidas entre 2 y 38 años, y en 6 pacientes existía historia familiar y la mayoría de ellos (13 de 15) habían notado la aparición de las lesiones antes de los 20 años3.
Varios han sido los casos publicados como AQE y que en realidad parecen corresponder a una HFA7-12. A pesar de considerar la HFA como un subtipo de AQE, todavía existe controversia sobre la relación etiopatogénica de ambos procesos, pues la diferencia principal estriba en la afectación o no de las fibras elásticas2, 13, 14.
Casi todos los pacientes descritos de HFA son de raza negra y de sexo femenino. Existen casos familiares y, aunque se desconoce el tipo de herencia, parece que la mayoría de los casos son de herencia autosómica dominante.
Presentamos un caso de HTA en una mujer de raza negra, que constituye el segundo caso español descrito bajo dicho término.
DESCRIPCIÓN DEL CASO
Se trata de un mujer de 27 años, de raza negra, natural de Guinea Ecuatorial, trabajadora de hostelería, sin antecedentes personales ni familiares de interés. Consultó por lesiones asintomáticas que se había visto hace dos años en manos y pies. En la exploración física se obser-vaban múltiples lesiones papulosas, de 1 a 2 mm, crateriformes (con una leve depresión central), agrupadas en caras laterales de pies, en regiones submaleolares y en caras laterales de la eminencia tenar de manos, así como lesiones aisladas en pliegues ventrales de las muñecas y en pliegues palmares (fig. 1). No presentaba lesiones en dorso de manos ni dedos, ni en pliegues plantares. No se detectaron lesiones ni alteraciones en pelo, uñas o dientes ni en otras localizaciones cutáneas. La paciente desconocía si existían familiares con lesiones similares. No presentaba historia de traumatismo crónico ni exposición a arsénico.
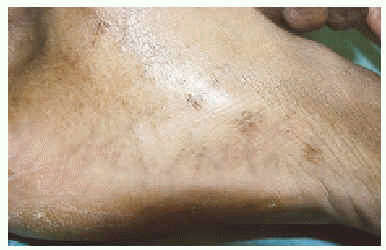
Fig. 1.--Múltiples lesiones papulosas crateriformes de 1 mm, agrupadas en cara lateral interna del pie izquierdo.
Se hicieron 4 biopsias: de región lateral del pie, de región submaleolar, de eminencia tenar y de pliegue palmar. Las 4 biopsias mostraban una imagen similar: hiperqueratosis sobre una epidermis ligeramente acantótica y deprimida, sin observarse daño actínico. Con tinción de Verhoeff-van Gieson y orceína no se demostró alteración de las fibras elásticas. En ninguna de las lesiones se observó imagen de columna de paraqueratosis, ni relación con el acrosiringio de glándula ecrina.
Las lesiones fueron tratadas con emolientes (urea al 10%), 5-fluorouracilo tópico y tretinoína al 0,025% tópica, sin que mostraran ninguna modificación.
DISCUSION
Varias son las patologías con una clínica similar, agrupadas bajo el término de acroqueratodermias papulosas marginales, consistentes en pápulas crateriformes a lo largo del borde de manos (con o sin afectación de pies), y que habría que incluir en el diagnóstico diferencial de la HFA. Algunos autores creen que todas ellas pudieran representar variedades de un mismo síndrome15, y proponen simplificar la clasificación en dos grupos: hereditarias (con y sin elastorrexis) y adquiridas16 (tabla 1).
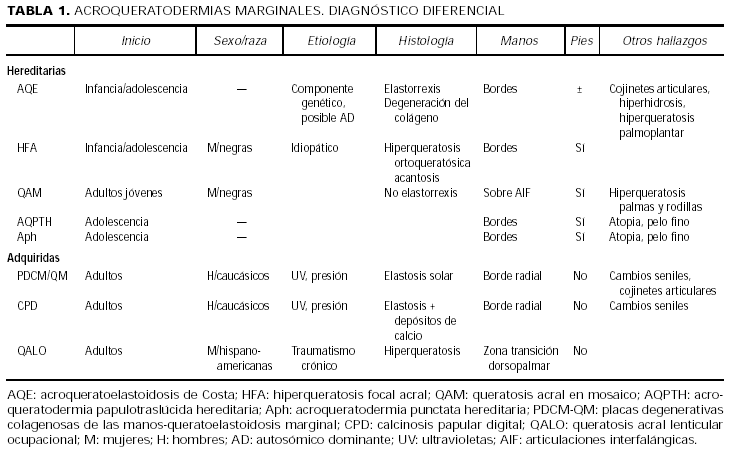
En el grupo de las hereditarias con elastorrexis se incluye a la AQE, y en el de las hereditarias sin elastorrexis la HFA, la queratosis acral en mosaico, la acroqueratodermia punctata hereditaria y la queratodermia papulotraslúcida hereditaria17. Algunos autores consideran estas últimas entidades variantes de la AQE clásica, pero sin elastorrexis ni alteraciones dérmicas4, 14, 15. Otros autores identifican la queratosis acral en mosaico con la HFA16.
Dentro del grupo de las adquiridas se describen:
1.Queratoelastoidosis marginalis18 o placas colagenosas degenerativas de las manos19. Ambos procesos parecen corresponder a la misma entidad. Aparecen normalmente en pacientes caucásicos de edad avanzada y con historia de exposición solar. Muestran una epidermis normal, con marcada degeneración actínica del colágeno y de las fibras elásticas.
2.Calcinosis papulosa digital, que difiere únicamente en la existencia de calcificaciones adyacentes a los fibroblastos20.
3.Queratosis acral lenticular ocupacional21, que aparece en bordes laterales de las manos de mujeres jóvenes expuestas al traumatismo crónico del lavado de ropa.
La aparición de lesiones en pliegues palmares obliga a hacer un diagnóstico diferencial con la queratodermia punteada de pliegues palmares. En esta entidad no aparecen lesiones en pies ni en caras laterales de manos y en la histología es típico ver una zona de hiperqueratosis centrada o relacionada con el acrosiringio de la glándula eccrina22, 23. Otros procesos que pueden conducir a un error diagnóstico con la HFA son las verrugas planas, la acroqueratosis verruciforme de Hopf, la poroqueratosis punctata y palmoplantar, el liquen plano palmar, el milium coloide, los xantomas palmares e incluso el elastoma juvenil5.
Las lesiones son normalmente asintomáticas, aunque han sido descritas asociadas a hiperhidrosis4 y a pseudoainhum24. Generalmente no es necesario realizar ningún tratamiento2, 4. Sin embargo, se ha intentado con el etretinato, con éxito relativo y recidiva de las lesiones tras la suspensión del fármaco8. La crioterapia puede ser una alternativa, pero no se han valorado resultados a largo plazo. También se han utilizado ácido salicílico, tretinoína16 y tazaroteno tópicos4.


