



Varón de 47 años de edad, que había sido estudiado en nuestro Servicio hacía 19 años por hiperpigmentación reticulada en el cuello, al igual que su hijo, y que no acudió a revisiones posteriores. Actualmente acudía a nuestra consulta remitido por el Servicio de Hematología, donde se encontraba en seguimiento por neutropenia y trombopenia. Su hijo había fallecido por una anemia aplásica severa.
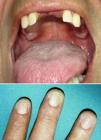
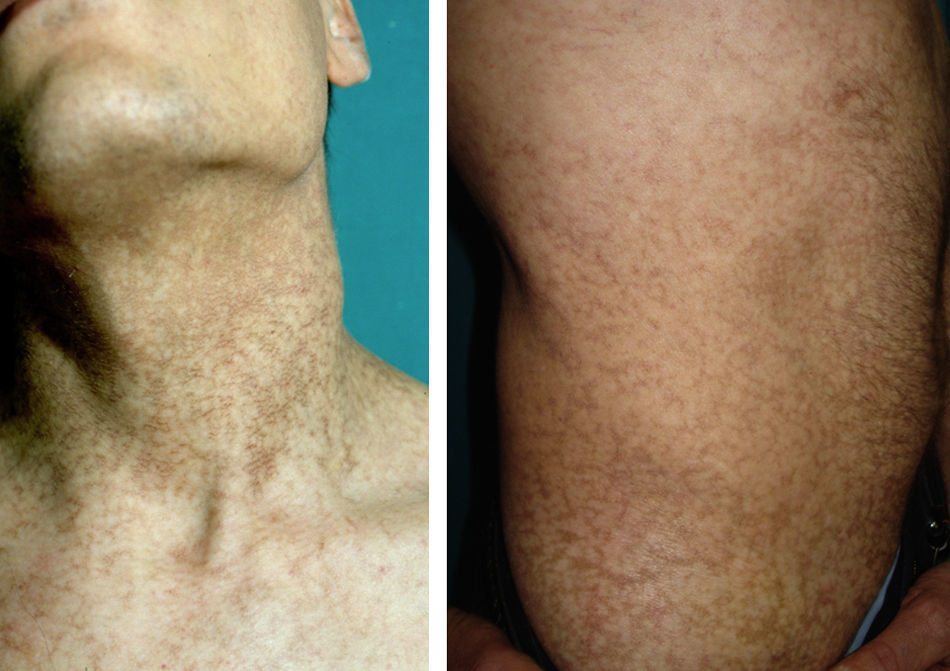
Exploración físicaA los 28 años el paciente presentaba una hiperpigmentación reticulada en el cuello y la porción superior del tronco, junto con hiperqueratosis e hiperhidrosis palmoplantar y distrofia ungueal (fig. 1). Su hijo (que entonces tenía 8 años de edad) también empezaba a desarrollar una alteración pigmentaria similar junto con distrofia ungueal.
En la actualidad la pigmentación reticulada del paciente se había extendido, había perdido múltiples piezas dentarias y presentaba una pequeña placa leucoplásica en la mucosa oral (fig. 2).
Histopatología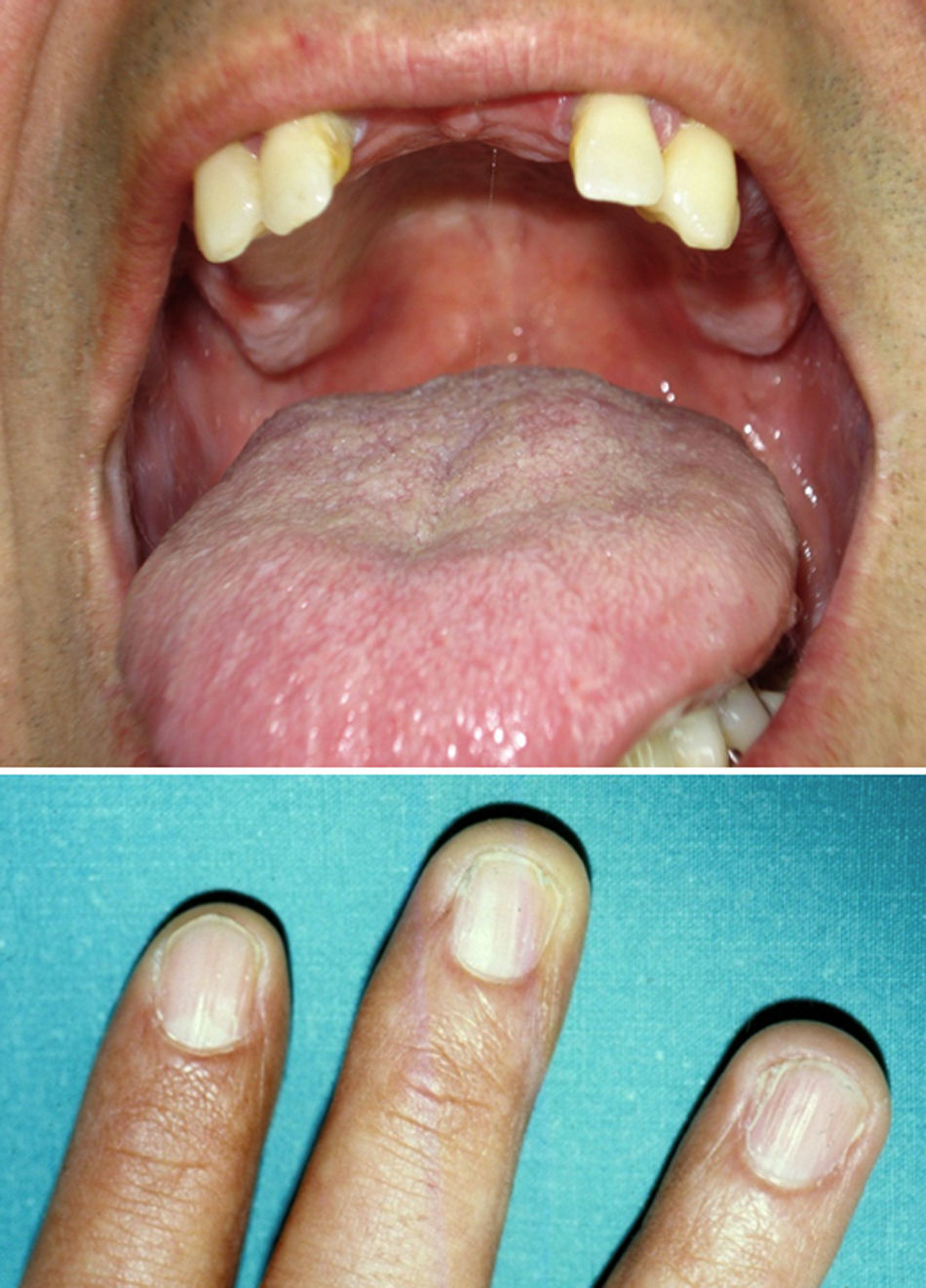
Una biopsia cutánea mostró una epidermis adelgazada junto con melanófagos en la dermis superficial.
Pruebas complementariasEn los análisis destacaba neutropenia y trombopenia. Una biopsia de médula ósea reveló una anemia aplásica grado ii con pérdida de serie megacariocítica y disminución de la granulocítica. El cariotipo de sangre periférica fue normal, y la estimulación con mitomicina C no produjo aumento de roturas cromosómicas.
¿Cuál es su diagnóstico?
DiagnósticoDisqueratosis congénita.
Evolución y tratamientoDesde el punto de vista dermatológico se ha recuperado el seguimiento periódico para la detección precoz de neoplasias malignas. El Servicio de Hematología lleva un seguimiento estrecho de su hemopatía y ha iniciado tratamiento con factores estimuladores de la trombopoyesis.
ComentarioLa disqueratosis congénita, también conocida como síndrome de Zinsser-Engman-Cole, es una genodermatosis con complicaciones multisistémicas graves que se caracteriza por pigmentación cutánea reticular, distrofia ungueal y leucoplasia1. El defecto molecular se encuentra en una alteración en el mantenimiento de los telómeros. La anomalía genética subyacente es heterogénea, habiéndose descrito varias mutaciones del complejo telomerasa2,3. Se han observado 3 patrones de herencia: recesiva ligada a X, autosómica dominante y autosómica recesiva, siendo la primera la más frecuente. En nuestro caso, los datos de los que disponemos (paciente varón, con un hijo varón afecto y ausencia de consanguinidad) sugieren una transmisión autosómica dominante. Con este patrón de herencia se ha descrito un fenómeno de anticipación por el que la clínica aparece de forma más precoz y más severa en sucesivas generaciones, en relación con un acortamiento progresivo de los telómeros4.
El pronóstico de los pacientes con disqueratosis congénita es malo. La insuficiencia medular (que ocurre en un 50% de los casos) y la predisposición al desarrollo de neoplasias malignas (sobre todo carcinomas epidermoides sobre áreas de leucoplasia) son las principales causas de muerte precoz en estos pacientes5. Aparte de los ya mencionados, se han descrito otros muchos hallazgos clínicos: hiperqueratosis palmoplantar, hiperhidrosis, canicie precoz, epífora, caries y pérdida de piezas dentales, retraso mental, talla baja, alteraciones pulmonares, fibrosis hepática, etc.1,2,5.
El diagnóstico diferencial debe hacerse con la anemia de Fanconi, el síndrome de Naegeli-Franceschetti-Jadassohn y la dermatopatía pigmentosa reticulada6. La anemia de Fanconi también muestra pancitopenia, aumento de neoplasias y alteraciones pigmentarias; sin embargo se trata de una hipermelanosis más difusa y se acompaña de anomalías óseas y un aumento de roturas cromosómicas al estimular los linfocitos con mitomicina C. El síndrome de Naegeli-Franceschetti-Jadassohn carece de leucoplasia y de afectación de médula ósea, y la hiperpigmentación reticulada desaparece durante la adolescencia. La dermatopatía pigmentosa reticulada, aunque también tiene hiperpigmentación y onicodistrofia, se caracteriza por la presencia de alopecia no cicatricial y no asocia afectación medular.
Se recomienda un abordaje multidisciplinar para tratar las complicaciones que pueden desarrollar estos pacientes. Desde el punto de vista dermatológico es necesario un seguimiento estrecho para detectar de forma precoz la aparición de carcinomas epidermoides sobre las áreas de leucoplasia6.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.


