
INTRODUCCION
Desde la descripción inicial por Smookler y Enzinger1 en 1982 se han recogido menos de 100 casos de fibroblastoma de células gigantes en la literatura médica. Cada vez son más los casos que se diagnostican debido a un mejor conocimiento de las características clínicas e histológicas de este tumor, que en el pasado era interpretado como sarcoma de bajo grado. Actualmente se conoce que se trata de un tumor mesenquimal benigno, localmente agresivo y con una alta tendencia a la recidiva local.
Descripción del caso
Un varón de 6 años de edad, sin antecedentes personales de interés, fue visto en el servicio de Cirugía Plástica al año y medio de vida, por presentar una lesión subcutánea en ingle izquierda. Se realizó una biopsia escisional de la misma para estudio histopatológico, observándose una proliferación mesenquimal compatible con fibromatosis. A los 6 años de edad, consultó de nuevo por la aparición de una lesión sobre la cicatriz quirúrgica, y se practicó una exéresis en bloque. El estudio histopatológico en esta ocasión fue de fibroblastoma de células gigantes, con márgenes libres. Dos meses después de esta intervención fue remitido a nuestro servicio por presentar una tumoración sobre la cicatriz quirúrgica, sugerente de queloide.
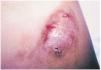
A la exploración física se observaba una tumoración eritematoviolácea de superficie abollonada y tacto fibroso, de aproximadamente 6 ×4 cm, sobre una cicatriz quirúrgica (fig. 1). Se tomó una biopsia en sacabocados de uno de los márgenes, que fue interpretada como recidiva local de FCG. El niño fue intervenido mediante extirpación amplia y autoinjerto.
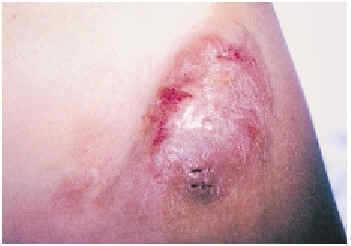
Fig. 1.--Tumoración nodular eritematopardusca, adherida a piel y planos profundos.
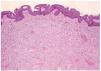
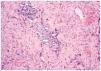
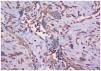
En el estudio histopatológico de la pieza se observó una lesión tumoral mal delimitada que ocupaba prácticamente toda la dermis (fig. 2) y que se extendía hacia el tejido celular subcutáneo. El tumor estaba constituido por células fusiformes y ovales, otras grandes de núcleos pleomórficos y células gigantes multinucleadas, en una estroma hialina (fig. 3). Se observaban además hendiduras, delimitadas por células grandes de núcleos hipercromáticos y células gigantes multinucleadas, que en su interior contenían células necróticas flotando en una sustancia basófila (fig. 4).
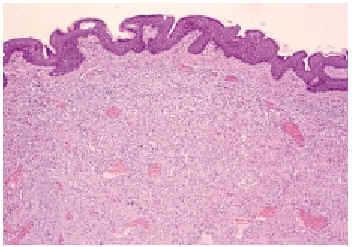
Fig. 2.--Tumoración bien vascularizada, constituida por células fusiformes que se extienden por toda la dermis reticular (hematoxilina-eosina, ×4).
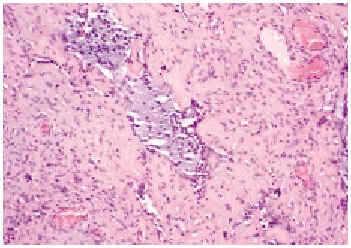
Fig. 3.--Se aprecian células de núcleos hipercromáticos en una estroma hialina, y seudoluces vasculares o «espacios angiectoides» con células necróticas en su interior (hematoxilina-eosina, ×20).
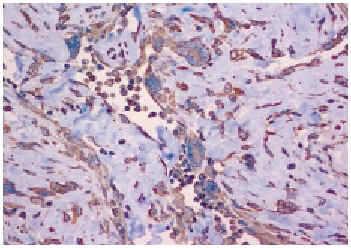
Fig. 4.--Tinción con vimentina fuertemente positiva. Se observan espacios angiectoides delimitados por células gigantes multinucleadas, rellenos de células necróticas (vimentina, ×20).
El estudio inmunohistoquímico demostró que las células tumorales expresaban vimentina, CD34 y CD68, y eran negativas para S-100, EMA, actina y desmina.
A los 8 meses se produjo una nueva recidiva local por lo que el paciente fue reintervenido en el Servicio de Cirugía Plástica. Dos años después de la última intervención está libre de enfermedad.
DISCUSION
El fibroblastoma de células gigantes es un tumor mesenquimatoso benigno de origen incierto, con alta tendencia a la recidiva local. Es un tumor poco frecuente que, generalmente, se presenta en las dos primeras décadas de la vida y muestra cierta predilección por afectar al sexo masculino (3:1)1. Clínicamente, se presenta como una lesión de superficie lisa, solitaria e indolora, de consistencia firme a veces blanda e incluso quística, que puede alcanzar hasta 7 cm de diámetro2. Tiende a localizarse en muslos, ingles y cara anterior del tórax3.
En el estudio histopatológico se observa una lesión tumoral asimétrica mal delimitada, localizada en dermis y/o tejido celular subcutáneo. El tumor está constituido por células fusiformes, con núcleos prominentes, dispuestas al azar en una estroma fibromixoide o hialina bien vascularizada, en la que, además, pueden observarse seudoluces vasculares o espacios angiectoides. Algunos autores sugieren que estos espacios se forman por dilatación progresiva de las hendiduras existentes entre el colágeno4; otros proponen que se forman tras un proceso de necrosis de algunas células tumorales5. Los hallazgos histológicos observados en nuestro caso apoyan esta última tesis, ya que pudimos comprobar la existencia de pequeñas acumulaciones de células grandes y células gigantes multinucleadas que sufrían un proceso de necrosis, para originar la formación de espacios angiectoides delimitados por células viables con células necróticas en su interior.
La presencia de células grandes de núcleos pleomórficos puede inducir a un diagnóstico erróneo de histiocitoma fibroso maligno o liposarcoma4,6. Ambos tumores son excepcionales en la infancia, suelen afectar a planos más profundos y presentan además una serie de rasgos histológicos que los diferencian. En el histiocitoma fibroso maligno hay mayor polimorfismo, mitosis atípicas y atipia celular, y en el liposarcoma existen lipoblastos.
El origen celular del fibroblastoma de células gigantes es un tema controvertido. El estudio inmunohistoquímico resulta positivo para vimentina y CD34 y negativo para actina, desmina, factor VIII y S-1007,8. En nuestro caso la expresión positiva de vimentina, CD34 y CD68 aboga a favor de un origen fibrohistiocitario.
Tras la descripción de casos de fibroblastoma de células gigantes que recurrían como dermatofibrosarcoma protuberans9 y viceversa10, y de lesiones híbridas en las que coexistían ambos tumores11,12, muchos autores han postulado la existencia de una relación histogénica entre ambos procesos. Para algunos, el fibroblastoma de células gigantes es una variante juvenil del dermatofibrosarcoma protuberans4. Para otros, ambas entidades representan distintos grados de diferenciación tumoral con distintas implicaciones biológicas10. Ambos tumores comparten una serie de rasgos clínicos, como la afectación de dermis y tejido celular subcutáneo, la tendencia a localizarse en tronco y el alto porcentaje de recidiva local e inmunohistoquímicos, ya que ambos son CD34 positivo11. Además, estudios citogenéticos recientes han puesto además de manifiesto la presencia de numerosos cromosomas en anillo con la translocación t(17,22)(q22;q13) en ambos tumores7,8.
Este defecto molecular da lugar a la fusión del gen que codifica para el colágeno tipo I A1 (COLIA1) localizado en el cromosoma 17 con el gen que codifica para la cadena beta del factor de crecimiento derivado de las plaquetas (PDGFB) localizado en el cromosoma 2213. Esta fusión hace que el gen PDGFB esté bajo el control del COLIA1, lo que produce la sobreexpresión de una proteína quimérica que actúa como PDGFB maduro. El dermatofibrosarcoma protuberans no sólo produce PDGFB sino que también posee receptores para el mismo, configurándose de este modo un circuito autocrino14,15 en el que las células tumorales estimulan su propio crecimiento, ya que la unión del PDGFB al receptor inhibe las señales de apoptosis celular15. Estos hallazgos proponen el bloqueo de estos receptores como nueva estrategia terapéutica15.
Aunque el origen celular del fibroblastoma de células gigantes y del dermatofibrosarcoma protuberans permanece oscuro, la relación temporospacial entre ambos, la frecuente expresión de CD34 y el hecho de que compartan el mismo defecto molecular sugiere, en nuestra opinión, que son una misma entidad, siendo el fibroblastoma de células gigantes la versión benigna y el dermatofibrosarcoma protuberans la versión maligna, con capacidad para presentar cambios sarcomatosos y metástasis a distancia.
El tratamiento de este tumor es puramente quirúrgico. Dado el alto porcentaje de recidiva local (50 %), como ocurrió en nuestro caso, la exéresis debe ser amplia, recomendándose un margen de 3 cm desde el borde palpable del tumor16. Actualmente, la microcirugía de Mohs supone la técnica quirúrgica de elección, debido a la mala delimitación del mismo17,18.
Aunque la radioterapia no está indicada en el tratamiento del fibroblastoma de células gigantes, sí se utiliza en formas agresivas de dermatofibrosarcoma protuberans a dosis de 50-60 Gy, sobre todo para prevenir recurrencias en resecciones tumorales incompletas. Dada la estrecha relación que existe entre ambos tumores, algunos autores consideran que su uso puede extrapolarse al fibroblastoma de células gigantes19.
En conclusión, hemos presentado un nuevo caso de fibroblastoma de células gigantes, un tumor con alta tendencia a la recidiva local, que requiere una exéresis amplia. Queremos incidir en la importancia del conocimiento de esta entidad por parte del dermatopatólogo y del anatomopatólogo para evitar que estos tumores sean interpretados como sarcomas de alto grado con las implicaciones terapéuticas que ello conllevaría.


