


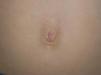


After Gaucher disease, Fabry disease is the most common storage disease caused by the progressive accumulation of glycosphingolipids in multiple organs. Clinical presentation is very varied, and furthermore, the manifestations in many of the organs affected are nonspecific.1 Diagnosis is thus challenging and often delayed. The average time from onset of symptoms to a diagnosis of Fabry disease is around 10 years.2 Cutaneous involvement is common and is one of the key signs that can lead a physician to suspect the disease. Skin manifestations include angiokeratomas, telangiectasias, abnormal sweating, and lymphedema. Recent studies of cutaneous involvement in Fabry disease have shown that the clinical spectrum of angiokeratomas is also varied.3 We performed a retrospective review of skin lesions in 5 patients (4 males and 1 female) with Fabry disease, with a particular focus on the clinical variants of angiokeratomas. The main clinical features are shown in Table 1. The majority of patients, including the only woman, had classic extracutaneous manifestations. The mean age at onset of angiokeratomas was 17.2 years. Patients #4 and #5 had a classic bathing trunk appearance, and patient #5 also had less hyperkeratotic vascular lesions on the palms of both hands. The other 3 patients had less characteristic angiokeratomas. Patient #1 had small, isolated lesions around the mouth and the umbilicus (Fig. 1), while patient #3 had extensive angiokeratomas distributed almost exclusively on the left side of the body. Finally, patient #2, who was heterozygous, had lesions in the form of angiokeratomas on the left side of the vulva (Fig. 2) and small isolated lesions on the anterior surface of the trunk. Just 2 of the patients had hypohidrosis. With the exception of patient #1, all the patients were receiving enzyme replacement treatment, with a mean uninterrupted treatment period of 9 years; there had been no signs of the angiokeratomas disappearing or becoming smaller or any improvements in the other skin manifestations.
Clinical Findings.
| Patient | Sex (M/F)/Age, y | Extracutaneous Manifestations | Location of Angiokeratomas | Age at Onset of Angiokeratomas, y | Duration of ERT, y | Other Skin Manifestations |
| 1 | M/11 | Exercise intoleranceCornea verticillata | Around mouth and umbilicus | 7 | No treatment | None |
| 2 | F/36 | AcroparesthesiaExercise intoleranceTinnitusDiarrheaCornea verticillata | Left lumbar region, vulva | 25 | 9 | Hypohidrosis |
| 3 | M/24 | AcroparesthesiaExercise intoleranceTinnitusAbdominal painCornea verticillata | Left body | 6 | 8 | None |
| 4 | M/50 | Left ventricular hypertrophyProteinuriaExercise intoleranceDiarrhea | Flank, waist, and medial aspect of thighs | 34 | 11 | Hypohidrosis |
| 5 | M/32 | AcroparesthesiaVascular tortuosity in both eyes | Thighs, buttocks, genitals, and palms | 14 | 10 | None |
Abbreviations: ERT, enzyme replacement therapy; F, female; M, male.
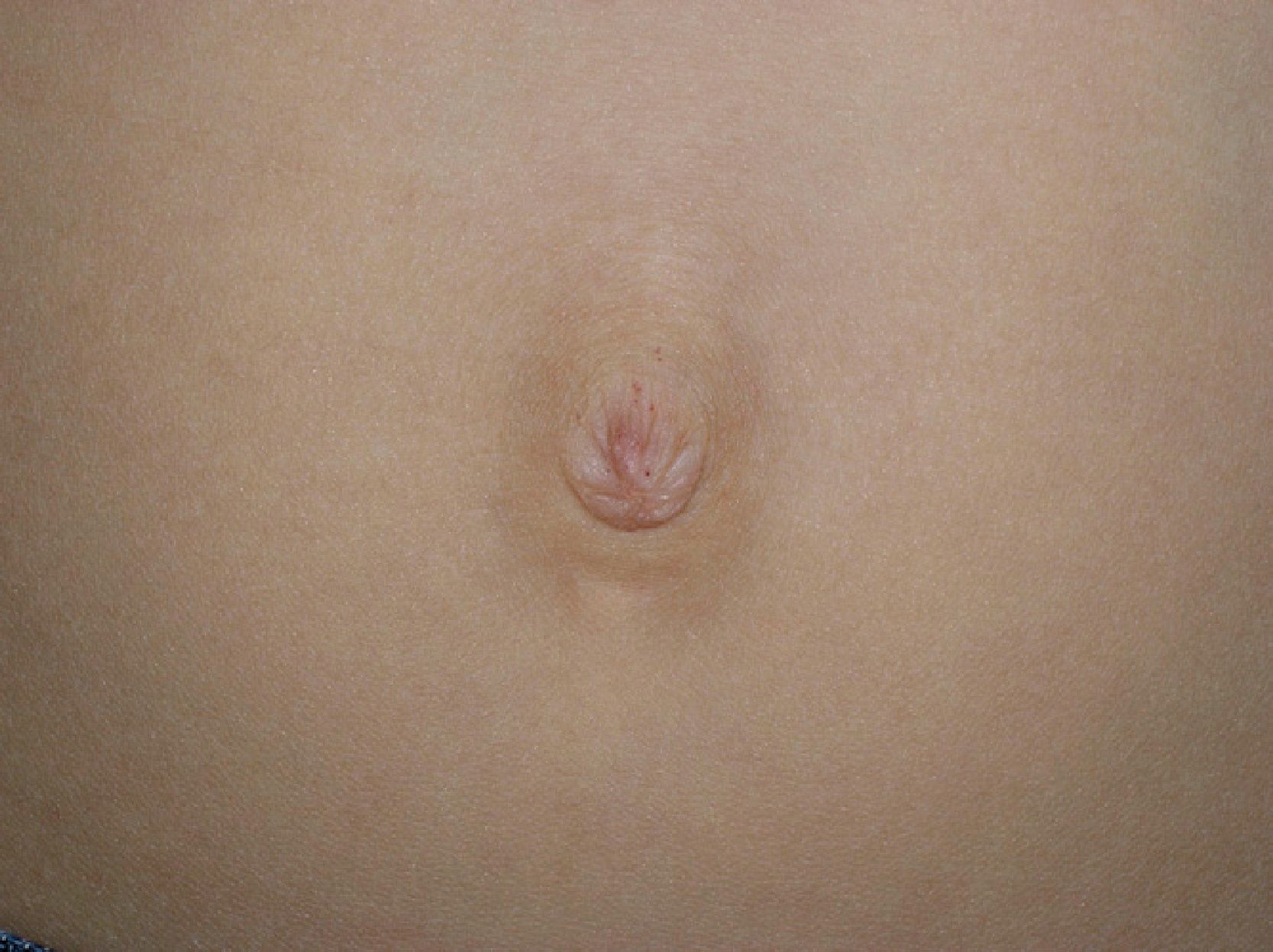

Angiokeratoma corporis diffusum is one of the 5 known types of angiokeratoma.4 It is highly characteristic of Fabry disease, but can also be seen in other lysosomal storage diseases.5,6 Angiokeratomas, which can appear in childhood or adulthood, develop in 66% of male patients with Fabry disease and in 36% of female patients.7 While the term angiokeratoma corporis diffusum suggests the presence of multiple angiokeratomas, this is not always the case. Furthermore, as seen in 3 of the 5 cases we describe, the lesions can appear in isolation or in clusters and can affect any part of the skin, mucous membranes, or genitals. Angiokeratomas classically occur in a bathing trunk distribution, referred to as such because the lesions typically affect the buttocks, the thighs, and the genitals. Of particular interest in our series was the unilateral, left-sided, distribution observed in 1 patient and the presence of small lesions in 2 patients (around the mouth and the umbilicus in one case and on the genital mucosa in the other). The detection of angiokeratomas on the genital mucosa can erroneously lead to a diagnosis of angiokeratoma of Fordyce.
The diagnosis of Fabry disease in a patient with lesions in less common sites is more difficult, but physicians should maintain a high level of clinical suspicion. While manifestations of the disease generally appear after the first decade of life, with acroparesthesia, angiokeratomas, exercise intolerance, and eye and gastrointestinal disorders, the main complications appear in young adults, who develop renal, cardiac, and cerebrovascular signs and symptoms. Considering that specific treatment is available for Fabry disease, early diagnosis (often based on the detection of angiokeratomas) is important, as timely treatment can prevent the development of serious, irreversible complications. Dermatologists can play a key role in this respect.
Enzyme replacement therapy (with agalsidase alpha) is the only specific treatment available for Fabry disease, and has been shown to stabilize renal function and reduce left ventricular hypertrophy.8–10 In our series, treatment did not improve any of the skin manifestations present before treatment initiation.
In conclusion, the detection of angiokeratomas, regardless of their location or distribution, should alert the dermatologist to a possible diagnosis of Fabry disease and prompt the performance of relevant tests to confirm or rule out this serious, treatable disease. Young relatives of newly diagnosed patients should also be studied, with a particular focus on symptoms that may have been missed or interpreted incorrectly.
Please cite this article as: Guinovart RM, et al. Enfermedad de Fabry: espectro clínico de los angioqueratomas. Actas Dermosifiliogr. 2013;104:261–3.


