







Introducción
En 1869 Nettleship y Tay describieron un desorden cutáneo que consistía en lesiones maculopapulosas pigmentadas con respuesta pruriginosa de tipo urticaria ante ciertos estímulos, como el rascado o el frotamiento de la piel, que fue llamado urticaria pigmentosa1. El descubrimiento por Paul Erlich en 1879 de las células mastocitarias demostró que dichas lesiones estaban formadas por cúmulos de mastocitos2.
Las mastocitosis reúnen un grupo de enfermedades caracterizadas por una anormal proliferación y acumulación de mastocitos en uno o más órganos, siendo la piel la localización más frecuentemente afectada. Los síntomas se desarrollan a partir de la infiltración de los órganos por los mastocitos y de la liberación de mediadores químicos mastocitarios.
Presentamos el caso de un paciente diagnosticado de mastocitosis sistémica, seguido durante largo tiempo en nuestro Servicio, cuya piel ha sufrido destacados y atípicos cambios clínicos durante la evolución.
Caso clínico
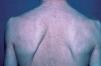
El paciente, varón de 75 años, fue visto por primera vez en nuestro Servicio de Dermatología hace 32 años por presentar la aparición progresiva, desde hacía 5 años, de lesiones papulonodulares amarillentas de predominio en el tronco (fig. 1), acompañadas de crisis ocasionales de prurito, taquicardia y flushing. No existían antecedentes patológicos dermatológicos personales ni familiares. El estudio analítico y la exploración sistémica fueron normales. La biopsia cutánea y tinción con hematoxilina-eosina mostró agregados dérmicos de células monomorfas redondeadas u ovales, con citoplasma eosinófilo; la tinción con azul de toluidina confirmó la naturaleza mastocitaria del infiltrado, por lo que fue diagnosticado de urticaria pigmentosa.
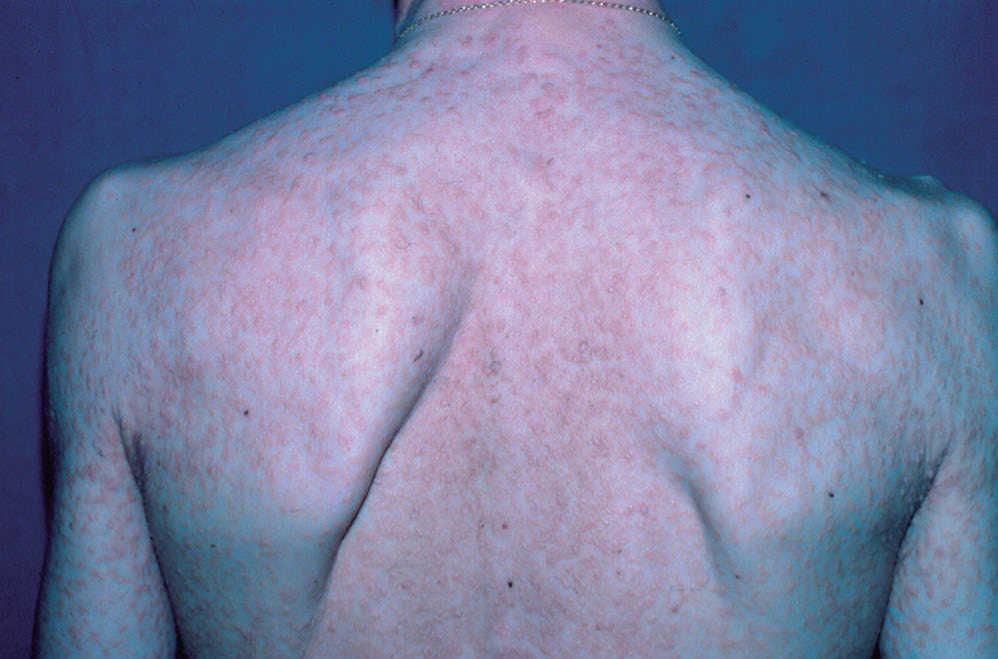
Figura 1. Lesiones cutáneas de nuestro paciente hace 32 años.
El paciente fue sometido a seguimiento con un completo estudio periódico, que ha incluido radiografías y gammagrafías óseas, pruebas de laboratorio, ecografía hepática, esplénica y biopsias cutáneas (todas fueron similares a la primera, con positividad para el azul de toluidina y el anticuerpo CD117, [fig. 2]). No se hallaron alteraciones significativas, hasta que en su última revisión detectamos una banda monoclonal IgG sérica, por lo que le fue practicado un aspirado y biopsia de médula ósea. El aspirado mostró una celularidad normal, pero en la biopsia aparecieron infiltrados multifocales densos, bien delimitados de mastocitos redondos típicos positivos para CD117, que constituye el patrón histológico más frecuente de afectación ósea en la mastocitosis sistémica3 (fig. 3). El nivel sérico de triptasa fue normal, 7,69 ng/ml (3-11 ng/ml).

Figura 2. Infiltrado de mastocitos en la dermis (tinción inmunohistoquímica con CD117, aumento original x100).
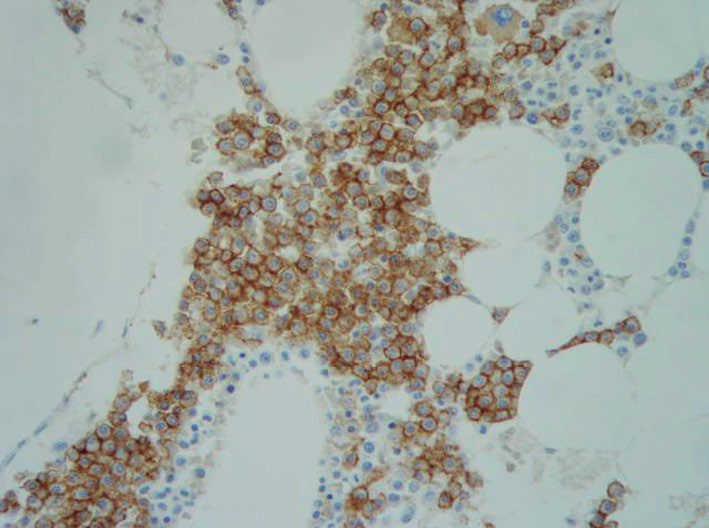
Figura 3. Biopsia de médula ósea (tinción inmunohistoquímica con CD117, aumento original x100).
El paciente fue también evaluado por el Servicio de Hematología, y diagnosticado de mastocitosis sistémica en fase indolente.
A lo largo de los años la piel ha sufrido cambios clínicos significativos; actualmente el paciente no presenta ninguna de las lesiones papulosas con las que se inició la enfermedad, sino que la piel ha adoptado un aspecto infiltrado, engrosado y paquidérmico de forma generalizada, con marcada acentuación de los pliegues cutáneos, que aparecen laxos y redundantes. También se han desarrollado numerosos comedones y tumores pediculados de tipo fibroma en la espalda, abdomen y regiones axilares e inguinales (figs. 4, 5 y 6). Los síntomas por liberación de mediadores mastocitarios han disminuido, por medio del tratamiento con antihistamínicos H1 (hidroxicina) y la evitación de los factores precipitantes.
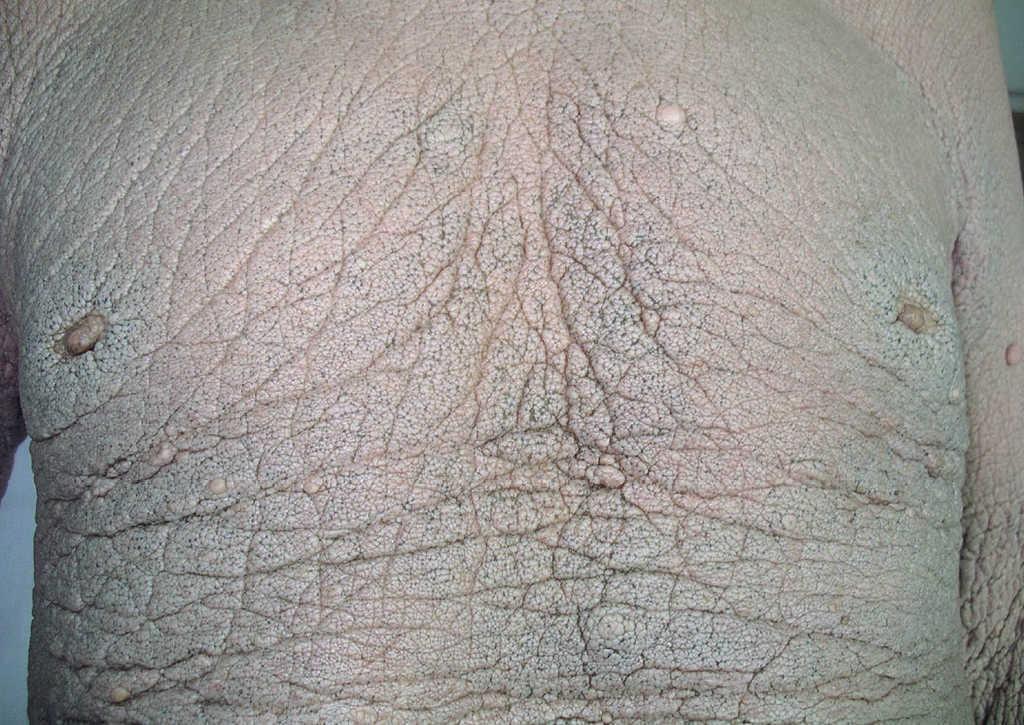
Figura 4. Piel grisácea, indurada y con numerosos comedones, que presenta el paciente actualmente.
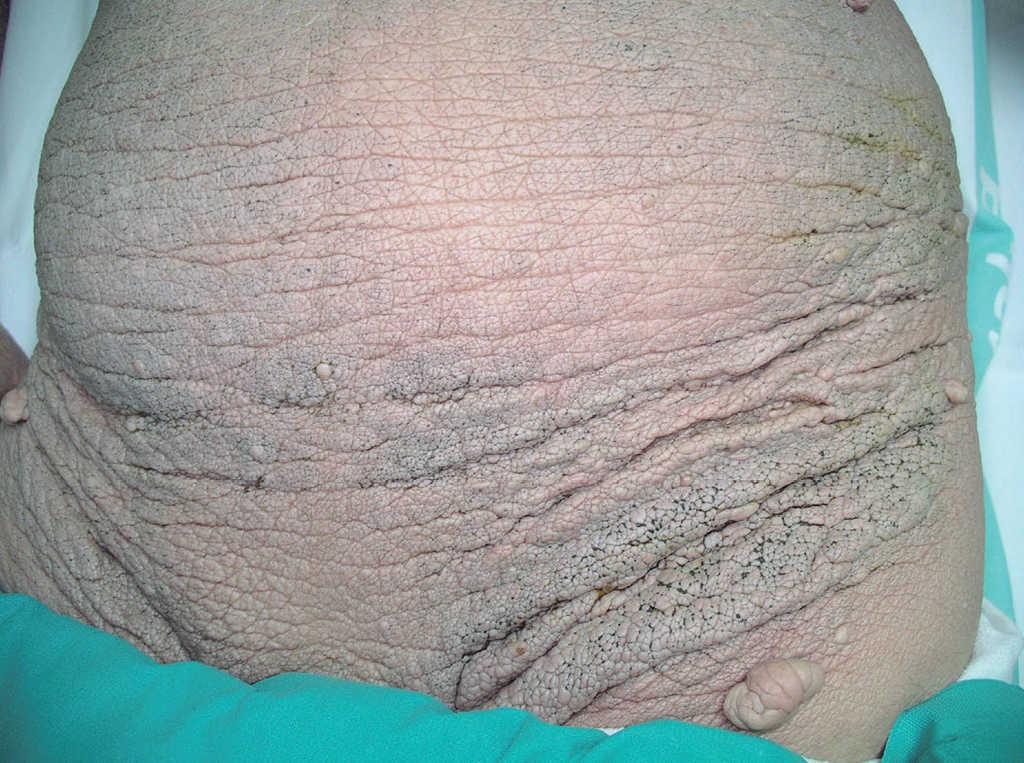
Figura 5. Aspecto similar de la piel del abdomen y regiones inguinales.

Figura 6. Lesiones pediculadaas tipo fibroma en la espalda del paciente.
Discusión
La mastocitosis sistémica se diagnostica más frecuentemente en los adultos, y viene definida por la infiltración multifocal de mastocitos en uno o más órganos, sin incluir la piel. La médula ósea se encuentra casi siempre afectada en estos casos.
De acuerdo con los recientes avances en la investigación sobre las mastocitosis, en 2001 se propuso una nueva clasificación de la Organización Mundial de la Salud (OMS) de estas enfermedades4. De forma general, las mastocitosis sistémicas se denominan agresivas o malignas cuando hay una proliferación incontrolada de mastocitos inmaduros en los órganos y/o circulación periférica, produciendo alteraciones funcionales, mientras que hablamos de formas indolentes cuando la enfermedad permanece estable, sin producir daño sobre los órganos.
En muchos casos pediátricos, y prácticamente en la totalidad de los adultos con mastocitosis sistémica indolente, las lesiones de la piel constituyen los únicos hallazgos clínicos5,6, sin hallarse otros signos debidos a la infiltración de los órganos por los mastocitos; de hecho, la morfología de estas células suele ser como las células maduras normales. La primera prueba que debe realizarse en los pacientes con sospecha de mastocitosis sistémica es la determinación de los niveles de triptasa sérica. En muchos casos de mastocitosis sistémica los valores de triptasa sérica se elevan a más de 20 ng/ml7,8. En cualquier caso, una determinación elevada de triptasa sérica, como único parámetro, no es definitiva para el diagnóstico de mastocitosis sistémica, puesto que estos valores también pueden elevarse en pacientes afectos de una neoplasia mieloide no mastocitaria, o en el curso de una reacción alérgica severa9.
En los pacientes adultos, incluyendo aquellos en los que la enfermedad se inició antes de la pubertad, el estudio de extensión debe incluir un aspirado y biopsia de médula ósea10,11; sin embargo, en la edad pediátrica el examen de médula ósea sólo debe realizarse si se detectan organomegalias, osteolisis o anomalías en el examen de sangre periférica.
Los pacientes con mastocitosis sistémica indolente suelen presentar lesiones de urticaria pigmentosa; muy raramente las lesiones están ausentes. Se ha descrito una forma extremadamente rara, multinodular no pigmentada, llamada variante pseudoxantomatosa, en la que las lesiones recuerdan al pseudoxantoma elástico12.
Nuestro paciente, en las primeras etapas de la enfermedad, presentó lesiones cutáneas de urticaria pigmentosa, pero a lo largo del tiempo la piel sufrió significativos cambios clínicos, que no fueron acompañados de diferencias desde el punto de vista histológico. Solamente hemos encontrado un caso similar recogido en la literatura (Meneghini y Angelini, 1980)13; su paciente, que sufría una mastocitosis sistémica, desarrolló una piel paquidérmica que compararon con el aspecto del cocodrilo, junto con comedones y fibromas. Paradójicamente, y al igual que en nuestro caso, el empeoramiento cutáneo fue unido a una mejoría de los síntomas y a una evolución benigna del cuadro sistémico.
Presentamos este caso por la rareza de las lesiones y la evolución atípica cutánea que ha sufrido nuestro paciente, que nosotros hemos nombrado «tipo paquidérmico» de mastocitosis. En la literatura sólo se ha descrito un caso similar; creemos que la piel paquidérmica podría ser secundaria a la presencia durante años de infiltrados mastocitarios cutáneos con liberación de mediadores, en una piel inicialmente afectada de urticaria pigmentosa.
Correspondencia:
María Pilar Sánchez-Salas.
Bario Mato, 38.
22314 Salas Altas. Huesca.
psanchezsalas@gmail.com
Aceptado el 15 de noviembre de 2006.


