



Historia clínica
Varón de 80 años sin antecedentes de interés, salvo gastritis crónica, que presenta hiperpigmentación marronácea progresiva en todo el tegumento, de cuatro meses de evolución acompañada de intenso prurito. El paciente había sido valorado dos años antes por unas lesiones en forma de pápulas y placas eritematosas de bordes netos en brazos, espalda e ingles. El estudio histológico en ese momento mostraba una dermatitis liquenoide avanzada sin ningún otro hallazgo específico.
Exploración física
A la exploración presentaba una hiperpigmentación marronácea generalizada, discretamente infiltrada a la palpación, que respetaba los pliegues abdominales, pliegues de piel del tórax e inguinales y descamación furfurácea generalizada (figs. 2 y 3). Muestra una hiperqueratosis palmo-plantar. La exploración general era normal, salvo la presencia de adenopatías axilares e inguinales bilaterales, simétricas, de aproximadamente 0,5 cm de diámetro, de consistencia elástica, no dolorosas ni adheridas a planos profundos.
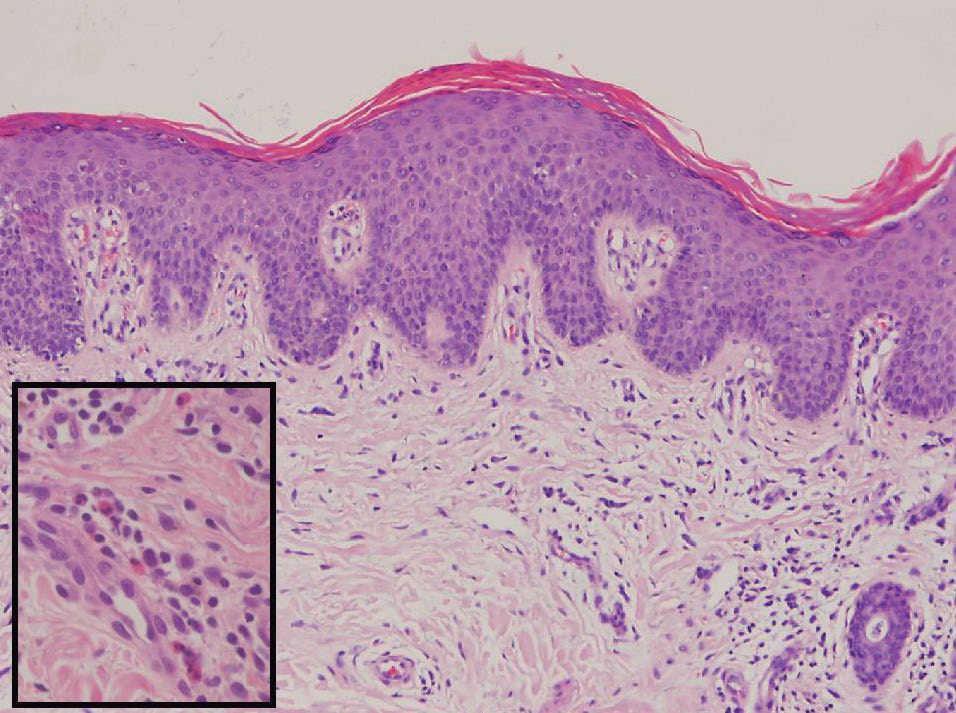
Figura 1. Hematoxilina-eosina, x30.

Figura 2.

Figura 3.
Exámenes complementarios
En la analítica se objetivó eosinofilia del 11 %, con el resto de la fórmula y cifra total de leucocitos normales. La lactacto deshidrogenasa (LDH) y la gammaglutamil transpeptidasa (GGT) estaban elevadas. Se realizó hemostasia, cuantificación de inmunoglobulinas, tomografía axial computarizada (TAC) body y ecografía abdominal con resultados normales. La serología para el virus de la inmunodeficiencia humana (VIH) fue negativa. Se solicitó inmunofenotipo en sangre periférica que fue normal, y no se encontraron células de Sèzary en la extensión de sangre.
Histopatología
La biopsia cutánea demostró una leve hiperplasia epidérmica con focos de espongiosis y fenómenos exudativos en la capa córnea, así como paraqueratosis. En la dermis superficial había un leve infiltrado inflamatorio linfocitario, con abundantes eosinófilos y cierta esclerosis de la dermis papilar.
¿Cuál es su diagnóstico?
Diagnóstico
Papuloeritrodermia de Ofuji.
Tratamiento y evolución posterior
Se pautó prednisona vía oral a dosis de 20 mg/día con buen control de la clínica y el prurito.
Comentario
La papuloeritrodermia de Ofuji (PO) fue descrita como un proceso clínico con entidad propia en 1984 por Ofuji et al1. Es una enfermedad de etiología desconocida que se caracteriza por la aparición de pápulas eritemato-marronáceas diseminadas, que confluyen hasta hacer una eritrodermia que característicamente respeta los grandes pliegues corporales (signo de la hamaca o deck chair sign)2.
La PO afecta con mayor frecuencia a varones que a mujeres, con una edad media de 60 años e incidencia aproximada de 1,5 por millón. Aparte de la afectación cutánea pueden existir otras alteraciones, tanto a nivel dermatológico (queratodermia palmoplantar e infartos ungueales) como en el plano analítico. El hallazgo de laboratorio más característico es la eosinofilia periférica. También pueden detectarse niveles altos de IgE y linfopenia. Las pruebas de función hepática pueden estar alteradas: aumento de la fosfatasa alcalina y GGT2.
La PO es en la mayoría de los casos idiopática. Se ha sugerido que es la expresión de enfermedades crónicas comunes de la piel, como eczema o psoriasis, en pacientes ancianos4. No obstante, la mayoría de los pacientes no tiene historia previa de enfermedad o problemas cutáneos. Se ha descrito, sin embargo, en un número importante de casos, la progresión a linfoma cutáneo de células T (LCCT), lo que sugiere que se pueda tratar de una dermatosis prelinfomatosa3. Este hecho justifica un control clínico e histológico estricto de estos pacientes.
La PO también se ha descrito asociada a neoplasias viscerales, linforma Hodgking, leucemia mieloide aguda, síndrome hipereosinófilo, sida, hipersensibilidad a medicamentos y sepsis biliar tras colecistectomía4,5.
La histología no es específica. La epidermis suele ser normal, pero puede encontrarse acantosis, espongiosis, paraqueratosis y ocasionalmente exocitosis4.
El diagnóstico se establece en función de la clínica, la analítica y la anatomía patológica.
Aunque con frecuencia la papulodermia es de causa desconocida, las asociaciones descritas en la literatura obligan al estudio y seguimiento de los pacientes.
Esto plantea la individualización del manejo de esta dermatosis poco común, que más bien debe considerarse una forma de eritrodermia que puede responder a diversas etiologías. El término PO debería usarse para casos como el de nuestro paciente, en que no se conoce la etiología de la dermatosis. El término papuloeritrodermia se limitaría a los casos con una etiología definida.
Por lo que respecta al tratamiento se han publicado en la literatura distintas opciones. Se obtiene mejoría con corticoides tópicos y sistémicos a dosis bajas, más antihistamínicos. También son efectivos el PUVA, UVB y re-PUVA6. Otras terapias como ciclosporina, etretinato, azatioprina e interferón alfa también han sido utilizadas con éxito6.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Montse Fernández Guarino.
Hospital Ramón y Cajal.
Carretera de Colmenar km 9,100. 28034 Madrid
montsefdez@msn.com
Aceptado el 3 de mayo de 2007.


