



Introducción
Los dermatofibromas son tumores dérmicos benignos que suelen aparecer en miembros inferiores, generalmente de mujeres jóvenes. La expresión «dermatofibromas eruptivos múltiples» (DFEM) se ha empleado clásicamente para definir la aparición de al menos 15 lesiones en pocos meses1. Recientemente se acepta que la aparición de 5 a 8 dermatofibromas en menos de 4 meses es más adecuado, ya que incluiría los casos incipientes2.
Los DFEM son una entidad poco frecuente, que se ha descrito asociada a pacientes con enfermedades autoinmunes, tratamiento inmunosupresor, neoplasias, trasplante de órganos e inmunodeficiencias (VIH)3.
Presentamos dos pacientes de 35 y 45 años con dermatofibromas eruptivos múltiples asociados a infección por el VIH y tratamiento inmunosupresor. Esta asociación es poco frecuente, y de hecho hemos hallado únicamente 10 casos descritos en la literatura. En uno de nuestros pacientes las lesiones aparecieron previamente al conocimiento de la enfermedad de base, lo cual es excepcional.
Casos clínicos
Caso 1
Mujer de 35 años natural de Santo Domingo sin antecedentes de interés que consultaba por la aparición de varias lesiones cutáneas asintomáticas e hiperpigmentadas, de tres meses de evolución, en la cara interna del muslo izquierdo y de forma aislada en la pierna derecha. Dichas lesiones fueron apareciendo de forma progresiva (fig. 1).
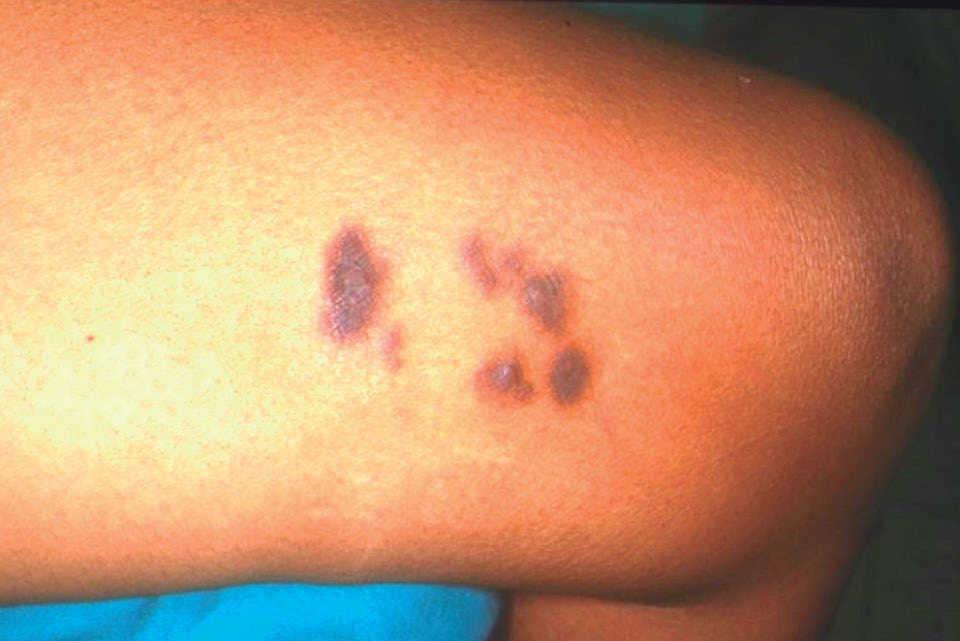
Figura 1. Nódulos eritematovioláceos en cara interna de muslo izquierdo, de 0,2-0,7 cm de diámetro, con cierta distribución arciforme y firmes a la palpación.
A la exploración se objetivaron un total de 7 nódulos eritematovioláceos que se agrupaban con distribución arciforme y tenían tamaños comprendidos entre 0,2 y 0,7 cm de diámetro, firmes a la palpación, en el muslo izquierdo, y una lesión similar de menor tamaño en la región posterointerna de la pierna derecha. La imagen dermatoscópica consistía en una estructura blanquecina central de bordes irregulares rodeada de un retículo estriado marrón claro.
El hemograma y bioquímica realizados no mostraron alteraciones, salvo una plaquetopenia periférica de 100.000 plaquetas/mm3 que no estaba presente en controles previos (el último hacía dos años). Debido a que la paciente no presentaba ninguna enfermedad sistémica aguda ni consumía fármacos que pudieran explicar el descenso de plaquetas mencionado, se realizó despistaje de infección por el VIH, dado que es una de las causas más frecuentes de plaquetopenia asintomática entre los 20 y 50 años. La positividad de dos ELISA se confirmó con Western-Blot. El número total de linfocitos CD4 fue de 500. Los anticuerpos antinucleares (ANA) resultaron negativos.
El examen histológico de una de las lesiones del muslo izquierdo mostraba un nódulo dérmico mal delimitado, formado por histiocitos y abundante proliferación de fibroblastos entre haces gruesos de colágeno. La epidermis suprayacente era acantósica y papilomatosa con cierta proliferación basaloide pigmentada. El estudio inmunohistoquímico fue positivo para factor XIIIa, confirmando la existencia de celularidad fibroblástica y negativo para S-100, desmina y actina.
Caso 2
Varón de 45 años, VIH conocido, en estadio B2 que recibía tratamiento antirretroviral desde hacía un año. Al ingreso el número de CD4 totales era de 250. El paciente se encontraba a cargo del Servicio de Gastroenterología debido a hemorragia digestiva alta, secundaria a varices esofágicas. Se solicitó valoración por el Servicio de Dermatología debido a la aparición de múltiples lesiones cutáneas en miembros inferiores, que sugerían un posible sarcoma de Kaposi. Además el paciente padecía cirrosis hepática secundaria a virus de la hepatitis C (VHC) y un carcinoma hepatocelular diagnosticado 6 meses antes de la aparición de las lesiones cutáneas, que fue tratado con capecitabina (profármaco del 5-fluorouracilo) dos meses antes de la consulta.
El paciente refería la aparición súbita de lesiones cutáneas localizadas en ambos miembros inferiores, que se iniciaron tras el tratamiento quimioterápico de forma progresiva.
En la exploración física se observaban 10 tumoraciones de color marrón rojizo y tamaños comprendidos entre 2 y 5 mm de diámetro, localizadas de forma bilateral en ambos miembros inferiores. En la región abdominal se observaban dos lesiones (fig. 2). Algunas mostraban una depresión característica de la piel suprayacente a la compresión lateral.
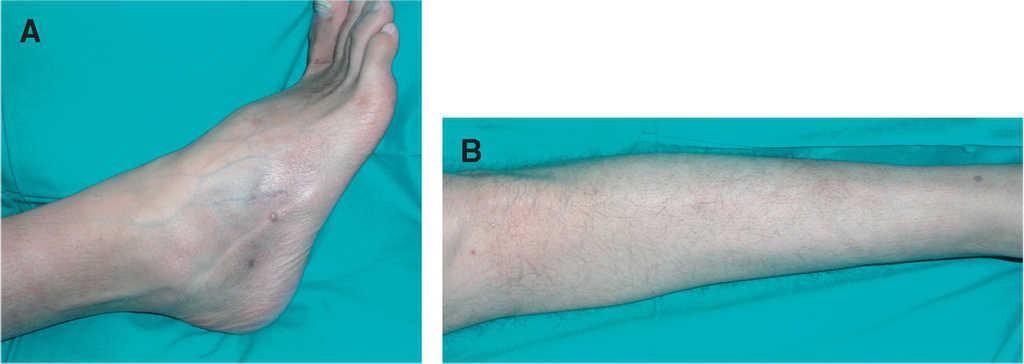
Figura 2. Pápulas firmes de coloración parda de 2-5 mm de diámetro diseminadas en tobillos (A) y área pretibial (B).
Los exámenes complementarios realizados, incluyendo hemograma, bioquímica y sistemático de orina, no revelaron datos adicionales.
La biopsia de una de las lesiones mostraba una histología e inmunohistoquímica muy similar a la del primer paciente, confirmándose el origen fibrohistocitario de las mismas.
Discusión
Los dermatofibromas son tumores dérmicos benignos frecuentes que suelen aparecer en miembros inferiores, generalmente de mujeres jóvenes. La expresión «dermatofibromas eruptivos múltiples» fue acuñada por Baraf y Shapiro en 1970, y se ha empleado clásicamente para definir la aparición de al menos 15 dermatofibromas1. Más recientemente se acepta que la aparición de 5 a 8 dermatofibromas en menos de 4 meses es más adecuado, ya que incluiría los casos incipientes, según sostienen Ammirati et al2.
La distribución de los DEFM suele predominar en extremidades inferiores, como ocurre en nuestros pacientes, pero pueden aparecer en otras localizaciones menos habituales, como extremidades superiores3 y región interescapular4.
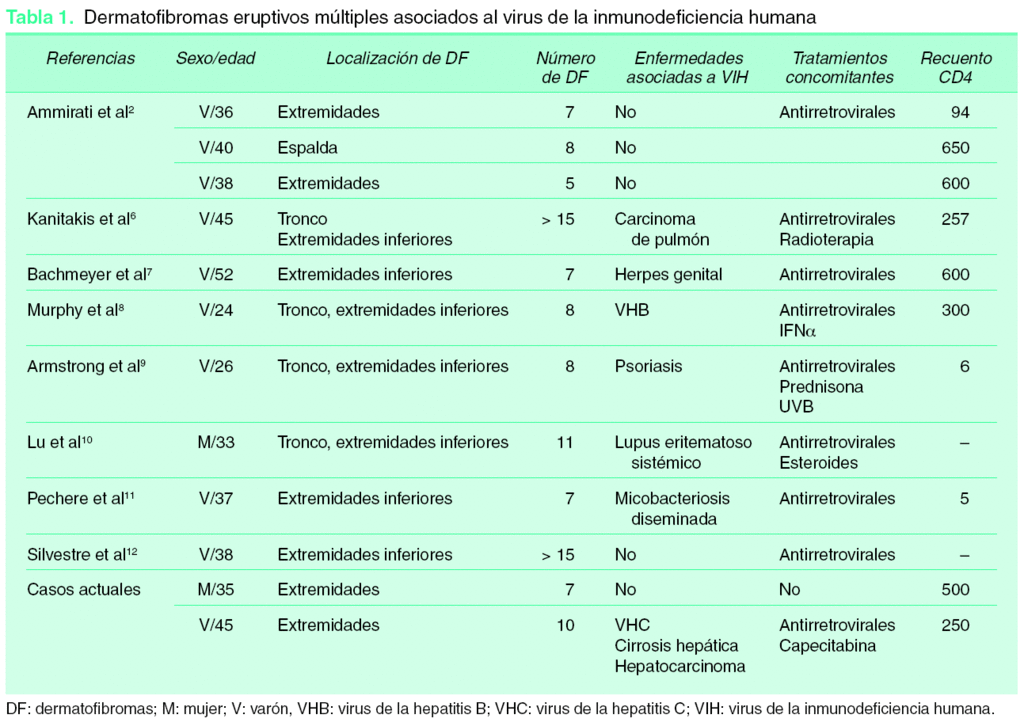
Los DFEM son más prevalentes en pacientes con enfermedades subyacentes5 (56 %). La mayoría de casos publicados se asocian a enfermedades autoinmunes en tratamiento con fármacos inmunosupresores (sobre todo lupus eritematoso sistémico, donde se han objetivado hasta un 46 % de casos). En segundo lugar los DFEM se asocian a infección VIH (32 %), como ocurre en los dos casos que presentamos, siendo en uno de ellos el diagnóstico de inmunosupresión posterior a la aparición de la clínica cutánea, lo cual es un hecho infrecuente, ya que en la inmensa mayoría de casos la inmunosupresión es conocida4. Hemos hallado 10 casos publicados de DFEM asociados al VIH2,6,-12, de los cuales 6 presentaban otras enfermedades graves concomitantes con posible influencia en la respuesta inmune. A esto hay que añadir la ingesta de fármacos inmunosupresores, además de la terapia antirretroviral, cuya introducción podría precipitar la clínica cutánea, como ocurre en uno de nuestros pacientes. En la literatura revisada no aparecen casos de VIH que comiencen con la aparición de dermatofibromas eruptivos (tabla 1).
Otras asociaciones documentadas son: obesidad, dislipidemias, embarazo y dermatitis atópica, pero dada la elevada prevalencia de algunas de estas patologías creemos que esta asociación no tiene excesivo valor.
Existen casos familiares y congénitos. También se han publicado casos asociados a colitis ulcerosa, leucemia mieloide aguda, leucemia mieloide crónica y trasplantes.
Recientemente se ha publicado el primer caso de DFEM asociados a síndrome de Down13.
En comparación con los DF clásicos, los DFEM presentan algunas características particulares desde el punto de vista clínico, pero no así desde el punto de vista histológico6. De este modo, en general los DEFM predominan en varones, se agrupan en racimos y se extienden al tronco con más frecuencia que los DF comunes.
La patogenia de los DF es aún poco conocida. Clásicamente se ha atribuido su aparición a un posible traumatismo o una picadura de insecto, por lo que sería esencialmente un proceso reactivo. Sin embargo, debido a su naturaleza persistente, en la actualidad se considera un proceso neoplásico benigno. Apoyando esta teoría se encuentran los trabajos de Yamamoto et al14,15 que han detectado alteraciones en la expresión de algunos factores de crecimiento fibroblástico. El hecho de que existan casos familiares refuerza la idea de que posiblemente haya un componente genético asociado16.
Se ha atribuido un papel destacado a las células mastocitarias en la patogenia de los DF17, dado que son fuente de citocinas que interfieren en la proliferación fibroblástica y epidérmica, así como en la migración linfocitaria. En los DFEM se ha detectado un mayor número de mastocitos en comparación con los DF solitarios. También se ha postulado que los DF pueden representar un proceso inmunorreactivo, mediado por las células dendríticas y otras células dérmicas18. De acuerdo con esto, el desarrollo de los DFEM en las inmunodeficiencias podría verse facilitado por el descenso en la población de linfocitos T. Algunos autores han sugerido que los DF asociados a VIH podrían ser formas atípicas de micobacteriosis12, lo cual no se ha demostrado. Karzakov et al determinaron la presencia de VHH-8 en los DFEM de un paciente VIH positivo mediante reacción en cadena de la polimerasa (PCR)19, pero este hallazgo se ha atribuido a la presencia en sangre del virus que puede existir en estos pacientes, ya que no se ha demostrado que exista replicación viral en las lesiones.
El diagnóstico diferencial debe realizarse principalmente con el dermatofibrosarcoma protuberans, con el que existen diferencias clínicas, histológicas e inmunohistoquímicas. También debe diferenciarse de otros tumores como leiomiomas, xantogranulomas y sarcoma de Kaposi.
Es posible que estas alteraciones cutáneas puedan ayudar al diagnóstico precoz de diversas enfermedades con afectación del sistema inmune, o a identificar una situación de mayor inmunosupresión.
Conflicto de intereses
Declaramos no tener ningún conflicto de intereses.
Correspondencia:
Cristina García-Millán.
Servicio de Dermatología.
Hospital Universitario Ramón y Cajal.
Carretera de Colmenar km. 9,100. 28034 Madrid.
cgmillan@gmail.com
Aceptado el 15 de noviembre de 2006.


