
INTRODUCCIÓN
Las eritroqueratodermias (EQ) son trastornos de la queratinizacion caracterizados por placas hiperqueratósicas eritematoanaranjadas con ausencia de lesiones palmoplantares y foliculares predominantes. En esencial se distinguen tres formas: EQ simétrica progresiva (EQSP) de Darier-Gottron (1), la EQ variable de Mendes da Costa (2) y un grupo heterogéneo de EQ atípicas con síntomas asociados como, por ejemplo, la EQ progresiva con sordera de Schnyder (3), la EQ en dianas de Dégos (4), la EQ localizada (5), la EQ con lesiones periorificiales (6) o la EQ anular migratoria (7). La EQSP es una rara genodermatosis de herencia autosómica dominante, con penetrancia incompleta y de expresividad variable, aunque hay casos de aparición espontánea (8). Fue descrita por primera vez en 1911 por Darier (9), aunque el nombre por el que le conoce hoy día se debe a Gottron (1). Desde su descripción inicial en 1911 se han recogido unos 40 casos en la literatura, incluyendo los 10 casos aportados por Ruiz-Maldonado y cols. en 1982 (10). En general, la EQSP muestra una afectación parcheada, que suele ser más o menos estable a lo largo de la vida o con tendencia a la mejoría, habitualmente hacia la pubertad. Nosotros presentamos un caso de EQSP parcheada que experimentó generalización hasta cubrir toda la superficie corporal, con un fenotipo idéntico a una eritrodermia ictiosiforme congénita no ampollosa.
DESCRIPCIÓN DEL CASO
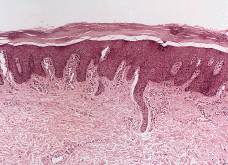
Un varón de 9 meses de edad fue remitido a nuestro Servicio en 1983 por presentar pocos meses después de su nacimiento unas placas rojizas y descamativas en la cara, extremidades y tronco que fueron intensificándose, acompañándose de afectación palmar sin otra sintomatologia acompañante. No existían antecedentes personales ni familiares de interés. En la exploración se apreciaban placas eritematoqueratósicas, bien delimitadas, simétricas y fijas, en ambas mejillas, en extremidades superiores, especialmente a lo largo de la zona de extensión, al igual que en extremidades inferiores (Fig. 1) y en parte del tronco. Presentaba, además, una leve queratodermia palmar. Se practicó analítica, incluyendo hemograma y bioquímica sanguínea, que resultaron dentro de los límites de la normalidad. Se realizaron dos biopsias de brazo y antebrazo que mostraron acantosis psoriasiforme con atrofia de la granulosa y capas alternantes horizontalmente de orto y paraqueratosis. Se veían ocasionales queratinocitos eosinófilos. En la dermis superficial había un ligero infiltrado linfohistiocitario perivascular (Fig. 2). Entre 1984 y 1993 se administró tratamiento con etretinato por vía oral a dosis de entre 0,5 y 1,5 mg/kg/día, con evidente mejoría en sus lesiones cutáneas. Las épocas de descanso del tratamiento se acompañaron de empeoramiento o reaparición de sus lesiones. La tolerancia clínica y analítica al tratamiento fue buena a lo largo de este período. En julio de 1993 el paciente dejó de seguir voluntariamente revisiones en nuestro Servicio.
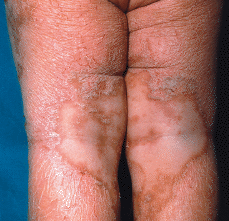
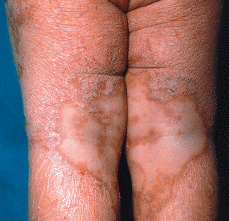
FIG. 1.--El paciente a la edad de 1 año. Placas eritematodescamativas anaranjadas con áreas de piel sana.
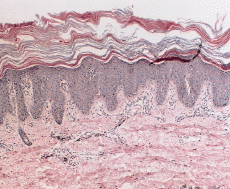
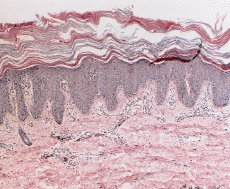
FIG. 2.--Biopsia cutánea tomada en el primer año de vida. Se aprecia acantosis psoriasiforme con atrofia de la granulosa y una córnea engrosada en la que alternan capas horizontales ortoqueratosis (azules) y paraqueratósicas (rojas). Leve infiltrado linfocitario en algunas papilas.
En enero de 2000, a los 17 años de edad, el paciente acudió de nuevo a la consulta presentando una generalización completa de sus lesiones hasta ocupar toda la superficie corporal. Este hecho había ocurrido de forma lenta y progresiva a lo largo de los últimos años. En la exploración, el paciente presentaba un aspecto eritrodérmico con descamación generalizada (Fig. 3), además de queratodermia palmoplantar con adelgazamiento de los dedos (Fig. 4) y ectropión. Se practicó una biospia cutánea que mostró una hiperqueratosis ortoqueratósica compacta con algunos focos de paraqueratosis y acantosis psoriasiforme con atrofia de la granulosa (Fig. 5). En la dermis se apreciaba una hiperplasia capilar superficial junto con un mínimo infiltrado linfohistiocitario perivascular. El paciente rehusó tratamiento con retinoides orales.
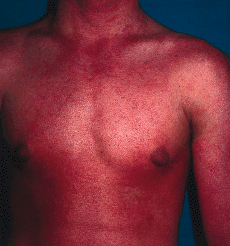
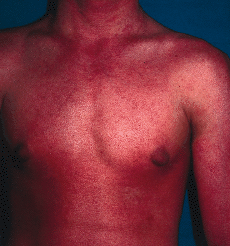
FIG: 3.--El paciente a los 17 años de edad. Eritrodermia generalizada con leve descamación.
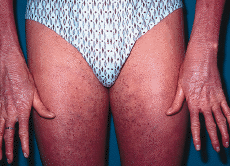
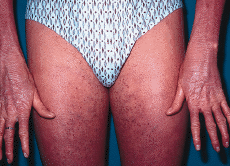
FIG. 4.--El paciente a los 17 años de edad. Eritema y leve descamación generalizada. Los dedos de las manos están afilados y rígidos.
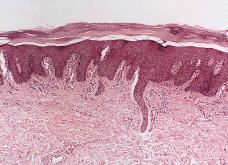
FIG. 5.--Biopsia cutánea tomada a los 17 años de edad. Existe hiperqueratosis ortoqueratósica compacta, atrofia de la granulosa con áreas de paraqueratosis, acantosis psoriasiforme e hiperplasia vascular dérmica superficial.
DISCUSIÓN
La EQSP se caracteriza clínicamente por placas hiperqueratósicas sobre una base eritematosa que se reconocen frecuentemente por un borde rojoanaranjado, bien delimitadas, simétricas y fijas, localizadas típicamente en extremidades, glúteos y cara (11-13). La afectación palmoplantar con queratodermia se ha descrito en el 50% de los casos. El tórax y el abdomen se afectan escasamente. Las primeras lesiones empiezan a aparecer durante el primer o primeros años de vida, con extensión lenta y progresiva de las lesiones durante los siguientes años hasta estabilizarse. La extensión e intensidad de las lesiones es variable y ocasionalmente hay mejoría o regresión espontánea hacia la pubertad (14). La distribución por sexos es similar. Se ha publicado una familia con EQSP que presentaba una mutación en el gen de la loricrina, consistente en la inserción de una citosina en posición 709, que origina un codon de terminación precoz de la cadena (15).
La afectación generalizada de toda la superficie corporal en pacientes con EQSP es excepcional, y tras una extensa búsqueda bibliográfica solamente hemos recogido un caso similar (16). Se trata de un niño afecto de EQSP que a la edad de 2 años y medio sufrió en un plazo de 2 meses un avance rápido de la enfermedad hacia una eritrodermia ictiosiforme, sin identificarse factores desencadenates del hecho. En dicho paciente los hallazgos histológicos y ultraestructurales fueron superponibles a los de la eritrodermia ictiosiforme congénita no ampollosa. En nuestro paciente los rasgos clínicos e histológicos fueron también similares a los de la eritrodermia ictiosiforme congénita no ampollosa, lo que resalta un cierto grado de superposición entre ambas enfermedades. Sin embargo, el defecto genético que subyace a la eritrodermia ictiosiforme congénita no ampollosa se encuentra en el gen de la transglutaminasa queratinocítica (17). Los estudios genéticos en un mayor número de familias con EQSP ayudarán a aclarar la situación nosológica de ésta.


